Standard Illumina sample decoding
The system uses a combination of two index segments to disambiguate samples, increasing the scaling capacity of sample pooling. Dual-indexing also creates longer barcodes to increase the fidelity of barcode decoding. The theoretical number of samples that can be pooled for sequencing is on the order of the product of the number of barcodes available for each index, however specific pairings that maximize differences between barcodes are encouraged by the arrangement of primers in some kit layouts.
Protocols
Library preparation kits that offer different barcode sets are available from multiple providers: Nextera, TruSeq, NEB, SMART-Seq2
Applications
Many common sequencing applications use standard dual-indexed barcoding. Examples include:
- Transcriptomics: bulk RNAseq, scRNA-seq
- Genome resequencing: Whole genome (WGS), whole exon (WES)
- Chromatin mapping/Epigenetics: ChIPseq, ATACseq, RIP-seq, CLiP-seq, PAR-CLIP
- Metagenomics and metatranscriptomics
- De novo genomics / transcriptomics
- Chromatin architecture: 3C, HiC
- Spatial transcriptomics
Read Anatomy
The final PCR products submitted for sequencing are composed as follows:
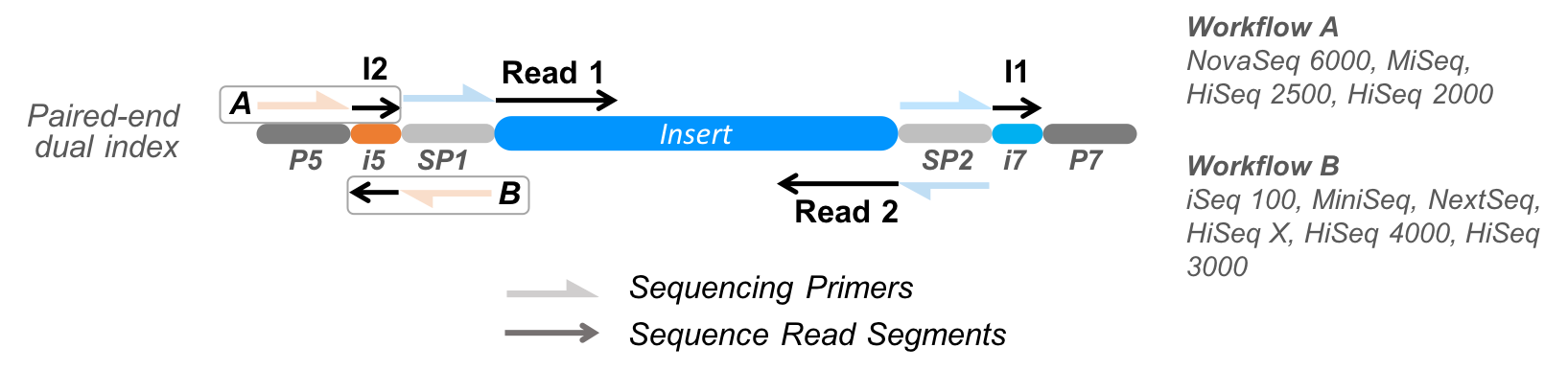
Per PCR product, there are two biological read segments (R1 and R2) and two index segments (I1 and I2). Read segments emerge from the sequencer in the following order: R1, I1, I2, R2. Notes:
- Different manufacturers use different adaptors for ligation (Pheniqs does not perform adapter trimming), as well as their own sets of standard index sequences for multiplexing.
- Pre-configured index libraries, or “codecs”, for most standard Illumina indexes are provided on the recipe page. Any configuration file can refer to these to “inherit” the pre-defined codecs.
- For dual-indexing, any i5-i7 pairings that are not used in the experiment should not be included in a fully specified configuration file.
- Different instruments read I2 on opposite strands (Workflow B), so reverse complemented i5 sequences must be entered into sample sheets if filling them out manually; if using run management software packages, these take care of reverse complementation automatically.
- The Pheniqs knit syntax can also reverse complement the extracted sequence before it is matched against the possible barcodes, so handling Workflow B instruments does not require users to manually reverse complement the sequences in the configuration file.
- Demultiplexed barcodes for both setups can be reported in either orientation depending on the configuration syntax used for barcode decoding. Pheniqs allows fine-grained control over this behavior, based on the desired output specified by the user.
Configuration file
This tutorial demonstrates how to manually prepare configuration files for decoding sample barcodes for a standard Illumina sequencing run. Pheniqs includes a Python API that helps users create configuration files automatically. For a tutorial that uses the Python API to generate the configuration files for this example, see the Standard Illumina sample decoding with the python API.
In this example the run has paired-end dual-index samples multiplexed using the standard Illumina i5 and i7 index protocol. The read is made of 4 segments: 2 biological sequences (cDNA, genomic DNA, etc.) read from both ends of the insert fragment, and 2 technical sequences containing the i5 and i7 indices. If the results are written to SAM, BAM or CRAM the sample barcode and its quality scores are written to the BC and QT tags, and the decoding error probability is written to the XB tag.
Input Read Layout
Base calling with bcl2fastq will produce 4 files per lane:
H7LT2DSXX_S1_L001_R1_001.fastq.gz: Read 1, starting from the beginning of the insert fragment (“top” strand).H7LT2DSXX_S1_L001_I1_001.fastq.gz: Index 1, the i7 index.H7LT2DSXX_S1_L001_I2_001.fastq.gz: Index 2, the i5 index.H7LT2DSXX_S1_L001_R2_001.fastq.gz: Read 2, starting from the other end of the insert fragment (“bottom” strand); note that this sequence is reverse complemented relative to the Read 1 sequence.
"input": [ "H7LT2DSXX_S1_L001_R1_001.fastq.gz", "H7LT2DSXX_S1_L001_I1_001.fastq.gz", "H7LT2DSXX_S1_L001_I2_001.fastq.gz", "H7LT2DSXX_S1_L001_R2_001.fastq.gz" ]declaring input read segments 2 biological and 2 technical sequences are often found in 4 FASTQ files produced by bcl2fastq base calling.
Specifying tokens for standard sequencing is very straight forward. We wish to extract the biological template sequences from Read 1 (R1) and Read 2 (R2) segments, the i7 index from the Index 1 (I1) read segment, and the i5 index from the Index 2 (I2) read segment. I1 and I2 may be 6, 8 or 10 nucleotides long, depending on the kit used. R1 and R2 may span as many bases as were sequenced, which will depend on the sequencing run configuration chosen (typically either 75 or 150nt for NextSeq and NovaSeq instruments), and may sometimes contain adapter sequences at their 3’ ends due to read-through of shorter inserts.
Token expression Segment index First Last Length Description 0::00end full R1: template (complete sequence) 3::30end full R2: template (complete sequence) 1::81078I1: i7 (8 nucleotides) 2::82078I2: i5 (8 nucleotides) Tokenization patterns for handling a standard Illumina paired-end dual indexed run with 8 nucleotide long indexes and variable length template segments.
To emit the two ends of the insert region as two segments of the output read, we declare the template transform
"template": { "transform": { "comment": "This global transform directive specifies the segments that will be written to output as the biological sequences of interest (here this is all of R1 and R2).", "token": [ "0::", "3::" ] } }declaring output read segments Only the segments coming from the first and the fourth file are biological sequences and should be included in the output. It is important to keep in mind that token coordinates are specified using Python array slicing syntax [start:end), with the start coordinate inclusive and the end coordinate exclusive. Notice that we specify the end coordinate on the tokens extracting the barcode segments. Some sequencing centers sequence I1 and I2 one nucleotide longer than necessary because that yields lower error rates on the last nucleotide. Explicitly specifying the end coordinates makes the configuration more robust to handle those cases and does not hurt otherwise.
To classify the reads by the i5 and i7 indices, we declare a decoder with a codec that lists the possible barcode sequences, along with any related metadata, and a transform that tells Pheniqs which read segment(s) and coordinates correspond to the barcode sequence(s).
"sample": { "transform": { "token": [ "1::8", "2::8" ] }, "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "barcode": [ "CGAGGCTG", "GTAAGGAG" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "barcode": [ "AAGAGGCA", "ACTGCATA" ] } } }declaring sample demultiplexing The standard Illumina dual-index protocol allows up to 96 unique dual 10 base barcodes in the i5 and i7 region, but for the sake of brevity we only show 2 here. Comments are allowed within any dictionary element in the configuration file and are ignored by Pheniqs. Alternatively, a codec may also be inherited from a base decoder rather than being enumerated explicitly here.
To discard reads that failed the internal Illumina sequencer noise filter, we instruct Pheniqs to filter incoming qc fail reads
"filter incoming qc fail": true
Putting things together, we can generate a configuration for demultiplexing the reads using the PAMLD decoder. Here, we declare that we expect 5% of reads to be foreign DNA (or noise), which should not classify to any of the barcodes (for instance spiked-in PhiX sequences). We specify the noise prior with the "noise": 0.05 directive. To mark as qc fail those reads with a posterior probability of correct barcode decoding that is below 0.95, we specify "confidence threshold": 0.95.
{ "PL": "ILLUMINA", "PM": "A00534", "filter incoming qc fail": true, "flowcell id": "H7LT2DSXX", "report url": "H7LT2DSXX_l01_sample_report.json", "input": [ "H7LT2DSXX_S1_L001_R1_001.fastq.gz", "H7LT2DSXX_S1_L001_I1_001.fastq.gz", "H7LT2DSXX_S1_L001_I2_001.fastq.gz", "H7LT2DSXX_S1_L001_R2_001.fastq.gz" ], "template": { "transform": { "token": [ "0::", "3::" ] } }, "sample": { "transform": { "token": [ "1::8", "2::8" ] }, "algorithm": "pamld", "confidence threshold": 0.95, "noise": 0.05, "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "barcode": [ "CGAGGCTG", "GTAAGGAG" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "barcode": [ "AAGAGGCA", "ACTGCATA" ] } } } }Single index Paired end Illumina protocol Classifying the 2 barcodes using the 4 fastq files produced by bcl2fastq for the first lane. For brevity, this is only an excerpt of the full configuration
Before we proceed we validate the configuration with Pheniqs:
pheniqs mux --config H7LT2DSXX_l01_sample.json --validate
The output is a readable description of all the explicit and implicit parameters after applying defaults. You can check how Pheniqs detects the input format, compression and layout as well as the output you can expect. In The Output transform section is a verbal description of how the output read segments will be assembled from the input. similarly Transform in the Mutliplex decoding section desribes how the segment that will be matched against the barcodes is assembled. the You can also see how each of the read groups will be tagged and the prior probability PAMLD will assume for each barcode. For brevity, the following is only an excerpt of the full validation report.
Environment
Base input URL .
Base output URL .
Platform ILLUMINA
Quality tracking disabled
Filter incoming QC failed reads enabled
Filter outgoing QC failed reads disabled
Input Phred offset 33
Output Phred offset 33
Leading segment index 0
Default output format sam
Default output compression unknown
Default output compression level 5
Feed buffer capacity 2048
Threads 8
Decoding threads 8
HTSLib threads 8
Input
Input segment cardinality 4
Input segment No.0 : ./H7LT2DSXX_S1_L001_R1_001.fastq.gz?format=fastq&compression=gz
Input segment No.1 : ./H7LT2DSXX_S1_L001_I1_001.fastq.gz?format=fastq&compression=gz
Input segment No.2 : ./H7LT2DSXX_S1_L001_I2_001.fastq.gz?format=fastq&compression=gz
Input segment No.3 : ./H7LT2DSXX_S1_L001_R2_001.fastq.gz?format=fastq&compression=gz
Input feed No.0
Type : fastq
Compression : gz
Resolution : 1
Phred offset : 33
Platform : ILLUMINA
Buffer capacity : 2048
URL : ./H7LT2DSXX_S1_L001_R1_001.fastq.gz?format=fastq&compression=gz
Input feed No.1
Type : fastq
Compression : gz
Resolution : 1
Phred offset : 33
Platform : ILLUMINA
Buffer capacity : 2048
URL : ./H7LT2DSXX_S1_L001_I1_001.fastq.gz?format=fastq&compression=gz
Input feed No.2
Type : fastq
Compression : gz
Resolution : 1
Phred offset : 33
Platform : ILLUMINA
Buffer capacity : 2048
URL : ./H7LT2DSXX_S1_L001_I2_001.fastq.gz?format=fastq&compression=gz
Input feed No.3
Type : fastq
Compression : gz
Resolution : 1
Phred offset : 33
Platform : ILLUMINA
Buffer capacity : 2048
URL : ./H7LT2DSXX_S1_L001_R2_001.fastq.gz?format=fastq&compression=gz
Output transform
Output segment cardinality 2
Token No.0
Length variable
Pattern 0::
Description cycles 0 to end of input segment 0
Token No.1
Length variable
Pattern 3::
Description cycles 0 to end of input segment 3
Assembly instruction
Append token 0 of input segment 0 to output segment 0
Append token 1 of input segment 3 to output segment 1
Sample decoding
Decoding algorithm pamld
Shannon bound 1 1
Noise 0.05
Confidence threshold 0.95
Segment cardinality 2
Nucleotide cardinality 16
Barcode segment length 8 8
Transform
Token No.0
Length 8
Pattern 1::8
Description cycles 0 to 8 of input segment 1
Token No.1
Length 8
Pattern 2::8
Description cycles 0 to 8 of input segment 2
Assembly instruction
Append token 0 of input segment 1 to output segment 0
Append token 1 of input segment 2 to output segment 1
Barcode undetermined
ID : H7LT2DSXX:undetermined
PU : H7LT2DSXX:undetermined
PL : ILLUMINA
PM : A00534
Segment No.0 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Segment No.1 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Barcode @A10_PDAC81
ID : H7LT2DSXX:CGAGGCTGGTAAGGAG
PU : H7LT2DSXX:CGAGGCTGGTAAGGAG
LB : A10_PDAC81
PL : ILLUMINA
PM : A00534
Concentration : 0.0101063829787234
Barcode : CGAGGCTG-GTAAGGAG
Segment No.0 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Segment No.1 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Barcode @A11_PDAC490
ID : H7LT2DSXX:AAGAGGCAACTGCATA
PU : H7LT2DSXX:AAGAGGCAACTGCATA
LB : A11_PDAC490
PL : ILLUMINA
PM : A00534
Concentration : 0.0101063829787234
Barcode : AAGAGGCA-ACTGCATA
Segment No.0 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Segment No.1 : ./H7LT2DSXX_l01.bam?format=bam&level=5
Output feed No.0
Type : bam
Compression : unknown@5
Resolution : 2
Phred offset : 33
Platform : ILLUMINA
Buffer capacity : 4096
URL : ./H7LT2DSXX_l01.bam?format=bam&level=5
pheniqs mux --config H7LT2DSXX_l01_sample.json --compile
While not strictly necessary, You may also examine the compiled configuration, which is the actual configuration Pheniqs will execute with all implicit and default parameters. This is an easy way to see exactly what Pheniqs will be doing and spotting any configuration errors.
Output format
By default Pheniqs will output uncompressed SAM to standard output. This allows you to quickly examine the output.
pheniqs mux --config CBJLFACXX_l01_sample.json|less
You can see Pheniqs will declare the annotated read groups and populate the BC, QT and XB tags.
@HD VN:1.0 SO:unknown GO:query
@RG ID:H7LT2DSXX:undetermined PL:ILLUMINA PM:A00534 PU:H7LT2DSXX:undetermined
@RG ID:H7LT2DSXX:CGAGGCTGGTAAGGAG BC:CGAGGCTG-GTAAGGAG LB:A10_PDAC81 PL:ILLUMINA PM:A00534 PU:H7LT2DSXX:CGAGGCTGGTAAGGAG
@RG ID:H7LT2DSXX:AAGAGGCAACTGCATA BC:AAGAGGCA-ACTGCATA LB:A11_PDAC490 PL:ILLUMINA PM:A00534 PU:H7LT2DSXX:AAGAGGCAACTGCATA
@PG ID:pheniqs PN:pheniqs CL:pheniqs mux --config H7LT2DSXX_l01_sample.json --output /dev/stdout --report /dev/stderr VN:2.0.6-546-g1178d4f6b8d044cd862150f44a88b01476b0e07d
A00534:24:H7LT2DSXX:1:1101:1018:1000 77 * 0 0 * * 0 0 ATATTTAAGAGGAGGCGTGGGGGCTGGCATAGGGCATCATTGAAGCCCTCTCCCTCTCCAATTAGTGGCCCCCTCTCCTCCCCTTTCCTTGGTTCAGCAGCAAGCAAAGAAAGAAAAAGAAATGTGGTCTCTTCCTCCTCCCTTCTTACTA ,FF,F:F,FFF,:FF::FF,:,:,F,F:,F:FF:FFF:FFFFF::,,,F:::::FFF,:FFFF,:F,:F:FF:,FFF,FFF,:,FFF,F,,:,::F,:,,F,,F:FFFFFFFF,:::,,,F,,,FFFFF:,,,F:,,FFFF:FF,,F,:,, RG:Z:H7LT2DSXX:TAGCGCTCGTAAGGAG BC:Z:TAGCGCTC-NTAAGGAG QT:Z:FFFFFFFF #:,FF::: XB:f:5.26752e-09
A00534:24:H7LT2DSXX:1:1101:1018:1000 141 * 0 0 * * 0 0 GATGAAGAATATAAAAGCCCATGAAGCCTTATTTCTTTTCCCTGCGTTTGAATGAAGAAGGGATGAGTAAGATACCACCTTTATTTTTATTTCTTTTCTTTAGTCTTAAAAAAGGAAAGTGTAGGATAGTGGTAAACTAATTGGAGATTGA ,,,:,F:,F,:,,FF:FF,F,F,,F,F,FF,F:FFF,FF,,F,,F,FF,,::,:FFF:FF,,:,,FF,:F,:,:FF:F,:F,,:F,F:,:FFFFF,,,:,,,:,,,::F:FFF:::F:::,,:F::,,,,:F,,,:F,FF,F,,::,,,F: RG:Z:H7LT2DSXX:TAGCGCTCGTAAGGAG BC:Z:TAGCGCTC-NTAAGGAG QT:Z:FFFFFFFF #:,FF::: XB:f:5.26752e-09
To write all multiplexed libraries into one BAM output file you can specify the output directive in the root of the configuration file
"output": [ "H7LT2DSXX_l01.bam" ]
Or directly on the command line
pheniqs mux --config H7LT2DSXX_l01_sample.json --output H7LT2DSXX_l01.bam
If you want to split the libraries and their segments into separate fastq files you can specify the output on the individual barcode directives. The additional instructions can be manually added or generated with the pheniqs-io-api.py by executing:
pheniqs-io-api.py -c H7LT2DSXX_l01_sample.json -LS -F fastq --compression gz > H7LT2DSXX_l01_sample_split.json
but briefly those are the necessery changes:
"sample": { "algorithm": "pamld", "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "barcode": [ "CGAGGCTG", "GTAAGGAG" ], "output": [ "H7LT2DSXX_l01_A10_PDAC81_s01.fastq.gz", "H7LT2DSXX_l01_A10_PDAC81_s02.fastq.gz" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "barcode": [ "AAGAGGCA", "ACTGCATA" ], "output": [ "H7LT2DSXX_l01_A11_PDAC490_s01.fastq.gz", "H7LT2DSXX_l01_A11_PDAC490_s02.fastq.gz" ] } }, "undetermined": { "output": [ "H7LT2DSXX_l01_undetermined_s01.fastq.gz", "H7LT2DSXX_l01_undetermined_s02.fastq.gz" ] } }Splitting the reads from different libraries To write the segments of the different libraries to fastq file specify the output on the individual barcode directives. Notice we explicitly declare where undetermined reads will be written, otherwise they will be written to the default output, declared at the root of the instruction. For brevity, the following is only an excerpt of the complete configuration for that scenario.
Statistics report
Once the run is complete, in addition to the output, a report is provied with statistics about the run and estimates for the prior that can be used in consecutive runs.
{ "incoming": { "count": 2894717093, "pf count": 2894717093, "pf fraction": 1.0 }, "sample": { "average classified confidence": 0.995714080516491, "average classified distance": 0.112188169938967, "average pf classified confidence": 0.999924673977143, "average pf classified distance": 0.093415423341184, "classified": [ { "average confidence": 0.993987748219644, "average distance": 0.145161376249895, "average pf confidence": 0.999873159581784, "average pf distance": 0.122156144917824, "barcode": [ "CGAGGCTG", "GTAAGGAG" ], "concentration": 0.010106382978723, "count": 33223879, "estimated concentration": 0.01145732888663, "index": 1, "low conditional confidence count": 986031, "low confidence count": 195561, "pf count": 33028318, "pf fraction": 0.994113842035121, "pf pooled classified fraction": 0.011645743335744, "pf pooled fraction": 0.011645743335744, "pooled classified fraction": 0.011665368351921, "pooled fraction": 0.011477418321929 } ], "classified count": 2848078003, "classified fraction": 0.983888204442229, "classified pf fraction": 0.995789089348196, "count": 2894717093, "estimated noise": 0.016178825488575, "index": 0, "low conditional confidence count": 46639090, "low confidence count": 11993002, "pf classified count": 2836085001, "pf classified fraction": 1.0, "pf count": 2836085001, "pf fraction": 0.979745139121959, "unclassified": { "count": 46639090, "index": 0, "pf count": 0, "pf fraction": 0.0, "pf pooled fraction": 0.0, "pooled classified fraction": 0.016375636464616, "pooled fraction": 0.01611179555777 } } }Partial statistics report
estimated concentrationandestimated noiseare the estimated priors.
Prior estimation
Better estimation of the prior distribution of the samples can improve accuracy. Pheniqs provides a simple estimation of the priors in the report from each run. Providing a file path with the--prior command line parameter in a prelimenary run will write new adjusted configuration file with concentration and noise values evaluated from the statistics collected from the run. The priors you specify in your initial configuration can be your best guess for the priors but you can simply leave them out altogether. Discarding output by specifying --output /dev/null on the prelimenary run can accelerate prior estimation.