- Pipeline overview
- Supported File Format
- Declaring Input
- Declaring Output
- Read transformation
- Output Manipulation
- Classifying by barcodes
- Providing Barcode Priors
- Minimum Distance Decoding
- Speeding things up
Pipeline overview
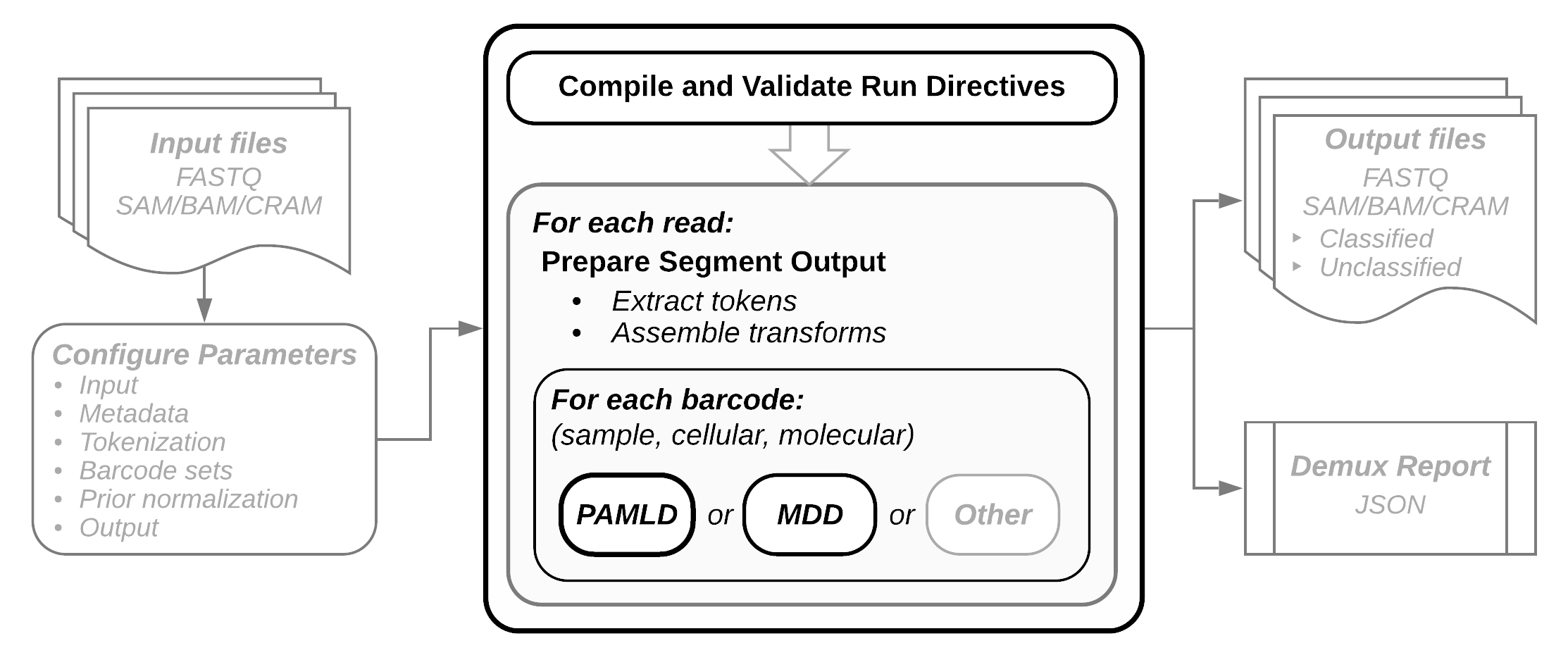
- Input: FASTQ or SAM formatted sequence files.
- Configuration: All runtime directives, including I/O, coordinate patterns identifying the location of sequence elements of interest, barcode sets, metadata, and any prior information about sample distributions. Trivial scenarios, such as format conversion or interleaving, can run with command line parameters only, but in most cases a configuration file will be required. Before execution begins the configuration file is compiled and validated. In the event of a validation failure the run is aborted and an informative error message is displayed.
- Tokenization: Input read segments are parsed to extract sequence element of interest. Output read segments are assembled as well as the sequence segmentsa that will be compared to the expected barcode instances.
- Decoding: Each barcode declared in the configation is decoded and potentially error corrected. Pheniqs performs either probabilistic decoding (PAMLD, preferred) or a simple minimum distance decoding (MDD). SAM metadata tags are populated with decoding results.
- Output: Biological sequences, observed and inferred barcode sequences, quality scores, and decoding error probabilities are emitted as output. Sequence Alignment/Map (SAM) format is preferred, but FASTQ may also be emitted.
- Run Report: Summary statistics about the decoding run are computed and written in a machine-readable JSON format, which can be easily parsed for visual display.
The Pheniqs command line interface accepts a JSON encoded configuration file containing directives for input and output layout, parsing read segments and run parameters. If you are new to JSON, a validator can be instrumental for troubleshooting syntax errors. Some parameters can also be specified as command line arguments that override their corresponding configuration file values. A brief description of the command line parameters Pheniqs accepts is always available with the -h/--help flags. If you use zsh the bundled command line completion will give you a more interactive command line experience.
Supported File Format
Pheniqs can arbitrarily manipulate reads from either SAM, BAM and CRAM or FASTQ. It can even operate on different read segments in different formats at the same time or interact with segments that are either interleaved into a single file or split over many. Using the intuitive addressing syntax you can classify reads by standard Illumina sample barcodes, multiple combinatorial cellular barcodes and molecular index tags in arbitrary locations along the read, all without pre or post processing the data.
Declaring Input
In this example we consider three files that contain synchronized segments from an Illumina MiSeq instrument. Pheniqs will construct the input read by reading one segment from each input file. Relative input and output file paths are resolved against the working directory which defaults to where you execute Pheniqs. You may optionally specify base input url to resolve relative paths and make the configuration file more portable.
For example we assume 3 FASTQ files created by executing bcl2fastq to simply get all 3 raw segments of a single indexed Illumina MiSeq run. The first four lines of each of the files are
000000000-BDGGG_S1_L001_R1_001.fastq.gz, the first read segment (or Read 1 in Illumina terminology):
@M02455:162:000000000-BDGGG:1:1101:10000:10630 1:N:0:
CTAAGAAATAGACCTAGCAGCTAAAAGAGGGTATCCTGAGCCTGTCTCTTA
+
CCCCCGGGFGGGAFDFGFGGFGFGFGGGGGGGDEFDFFGGFEFGCFEFGEG
000000000-BDGGG_S1_L001_I1_001.fastq.gz, the second read segment (or Index 1 in Illumina terminology):
@M02455:162:000000000-BDGGG:1:1101:10000:10630 2:N:0:
GGACTCCT
+
B@CCCFC<
000000000-BDGGG_S1_L001_R2_001.fastq.gz, the third read segment (or Read 2 in Illumina terminology):
@M02455:162:000000000-BDGGG:1:1101:10000:10630 3:N:0:
GCTCAGGATACCCTCTTTTAGCTGCTAGGTCTATTTCTTAGCTGTCTCTTA
+
CCCCCGGGGGGGGGGGGGGGGGGGF<FGGGGGGGGGGGGFGFGGGGGGGGG
To declare those files as input you add an input directive, which is a JSON array of file paths. The order of the paths in the array enumerates the input read segments so Pheniqs will expect to read one segment from each file you list. You may provide the input segments in any order you wish as long as you adhere to that order throughout the configuration file.
{ "input": [ "000000000-BDGGG_S1_L001_R1_001.fastq.gz", "000000000-BDGGG_S1_L001_I1_001.fastq.gz", "000000000-BDGGG_S1_L001_R2_001.fastq.gz" ] }Example 1.1 Declaring an input read that is split over three gzip compressed FASTQ files. Since the file paths do not start with
/they are considered relative and resolved against the current working directory. If you specifybase input urlpaths are resolved relative to that directory path. Absolute file paths ignorebase input url.
Notice that the order of the paths in the array is not just telling Pheniqs where to find the files but actually defines the enumerated segments of the input read. When reading the split read layout in this example that just means Pheniqs will read one segment from each input file. But if the same 3 segment input reads were interleaved into one file, you would list the same file path 3 times to tell Pheniqs that every 3 consecutive records in that file form a single sequence read with 3 segments.
Example 1.1 is already a complete and valid Pheniqs configuration file! Since we have not yet specified any output or manipulation instructions, reads are simply interleaved to the default stdout in SAM format. Executing it will yield the following output on stdout:
@HD VN:1.0 SO:unknown GO:query @RG ID:undetermined PU:undetermined M02455:162:000000000-BDGGG:1:1101:10000:10630 77 * 0 0 * * 0 0 CTAAGAAATAGACCTAGCAGCTAAAAGAGGGTATCCTGAGCCTGTCTCTTA CCCCCGGGFGGGAFDFGFGGFGFGFGGGGGGGDEFDFFGGFEFGCFEFGEG FI:i:1 TC:i:3 M02455:162:000000000-BDGGG:1:1101:10000:10630 13 * 0 0 * * 0 0 GGACTCCT B@CCCFC< FI:i:2 TC:i:3 M02455:162:000000000-BDGGG:1:1101:10000:10630 141 * 0 0 * * 0 0 GCTCAGGATACCCTCTTTTAGCTGCTAGGTCTATTTCTTAGCTGTCTCTTA CCCCCGGGGGGGGGGGGGGGGGGGF<FGGGGGGGGGGGGFGFGGGGGGGGG FI:i:3 TC:i:3Example 1.2 Output header and first 3 records (one complete read) from interleaving the three read segments verbatim into a single SAM formatted stream written to standard output using the configuration file in Example 1.1.
In the follwing example you will see how to use this simple example to interleave the raw split read segments into a single CRAM file. CRAM files are the latest indexed and compressed binary encoding of SAM implemented by HTSlib and often provide more efficient compression than the ubiquitous gzip compressed FASTQ format while being much faster to interact with. Packaging your reads into a CRAM container also makes archiving raw data simple. Another huge advantage of interleaved files is that they may be produced or consumed by Pheniqs through standard streams.
Declaring Output
Since in most cases you do not want output to be delivered to stdout, you will need to provide an output file path. To write interleaved output to a compressed CRAM file simply add an output directive to Example 1.1. Like input, the output directive is a JSON array of file paths. To interleave all output segments into the same file, specify only that one path in the output array. Alternatively, to split the output to multiple files, specify the same number as there are segments in the output read. You may optionally specify base output url to resolve relative paths and make the configuration file more portable. Absolute file paths ignore base output url.
{ "input": [ "000000000-BDGGG_S1_L001_R1_001.fastq.gz", "000000000-BDGGG_S1_L001_I1_001.fastq.gz", "000000000-BDGGG_S1_L001_R2_001.fastq.gz" ], "output": [ "000000000-BDGGG_raw.cram" ] }Example 1.3 Interleaving three read segments verbatim into a single CRAM file. CRAM and BAM files are often much faster to read and write, especially in highly parallelized environments, and also support a rich metadata vocabulary.
Before we move on to read manipulation here is the same interleaving configuration achieved without a configuration file by specifying the same instruction on the command lines
pheniqs mux \ --input BDGGG_s01.fastq.gz \ --input BDGGG_s02.fastq.gz \ --input BDGGG_s03.fastq.gz \ --output 000000000-BDGGG_raw.cramExample 1.3.1 Interleaving three read segments verbatim into a single CRAM file without a configuration file. Notice that
--inputis specified 3 times and the order the arguments are provided on the command line enumerates the input segments.
Read transformation
Pheniqs provides a generic method to derive new sets of sequence segments from the input read. A set can form the desired output or it can be a technical artifact used to classify the biological sequence. Either way, the syntax is the same. When declared inside the template section, the transform directive constructs the output read segments. When declared inside a barcode decoder, it constructs a segmented sequence that is assessed against the list of expected barcodes.
Transforms operate on the input read in two steps: First the token element, a JSON array of tokenization patterns, that each extract a continuous sequence (or a token) from an input segment, and second the knit element that constructs new segments from the previously defined tokens.
If the segments you are extracting can be found as one continuous sequence in a read segment you only need to specify the token array and Pheniqs will assume that each token represents a segment. If, however, you are trying to construct a segment from multiple non continuous tokens or need to reverse complement the sequence you have one more step. The optional knit directive references the tokens to construct a new segment from multiple tokens. If the knit array is omitted from transform, each token is assumed to declare a single segment.
Output Manipulation
The transform directive in the template section is used to manipulate the output read. If omitted all segments of the input are written verbatim to the output, as seen in Example 1.1 and Example 1.3. Since the second segment contains only a technical sequence, and we do not want to write it to the output, we add a transform directive to construct an output read from only the first and third segments of the input.
{ "input": [ "000000000-BDGGG_S1_L001_R1_001.fastq.gz", "000000000-BDGGG_S1_L001_I1_001.fastq.gz", "000000000-BDGGG_S1_L001_R2_001.fastq.gz" ], "template": { "transform": { "token": [ "0::", "2::" ] } } }Example 1.4 Adding a transform directive composing the output read from only the untouched first and third input segments. Input segments in Pheniqs are indexed and referenced using a zero based coordinate system so the first segment is 0 and the third is 2. Since we want to output the entire first and last segments the start and end coordinates in the pattern are left out to accept their default values.
A token patterns declared in the token array of the transform directive is made of 3 colon separated integers. The first is the zero based input segment index, as enumerated by the input array. The second is an inclusive zero based start coordinate to the beginning of the token and it defaults to 0 if omitted. The third is an exclusive zero based end coordinate to the end of the token. If the end coordinate is omitted the token spans to the end of the segment. The two colons are always mandatory. Pheniqs token pattern can address segments from either the 5’ (left) or 3’ (right) end. To address the 3’ end you use negative coordinates. Since token pattern coordinates mimic the python array slicing syntax they are fairly easy to test.
@HD VN:1.0 SO:unknown GO:query @RG ID:undetermined PU:undetermined M02455:162:000000000-BDGGG:1:1101:10000:10630 77 * 0 0 * * 0 0 CTAAGAAATAGACCTAGCAGCTAAAAGAGGGTATCCTGAGCCTGTCTCTTA CCCCCGGGFGGGAFDFGFGGFGFGFGGGGGGGDEFDFFGGFEFGCFEFGEG M02455:162:000000000-BDGGG:1:1101:10000:10630 141 * 0 0 * * 0 0 GCTCAGGATACCCTCTTTTAGCTGCTAGGTCTATTTCTTAGCTGTCTCTTA CCCCCGGGGGGGGGGGGGGGGGGGF<FGGGGGGGGGGGGFGFGGGGGGGGGExample 1.5 Output header and first 2 records (one complete read) from interleaving the three read segments to a SAM formatted stream using the configuration file in Example 1.4. Notice that only the first and third segments were written to the output.
Classifying by barcodes
The reads in our files were sequenced from DNA from 5 individually prepared libraries that were tagged with an 8bp technical sequence before they were pooled together for sequencing. To classify them by this sample barcode into read groups we need to examine the first 8 nucleotides of the second segment. If we were absolutely confident no errors occurred during sequencing decoding the barcode could be as trivial as comparing two strings. In a real world scenario however each nucleotide reported by a sequencing instrument is accompanied by an estimate of the probability the base was incorrectly called. We refer to such an uncertain sequence as an observed sequence. The sample directive is used to declare a decoder that will classify the reads by examining the segments constructed by the embedded transform and comparing them to the sequence segments declared in the codec element.
{ "sample": { "transform": { "token": [ "1::8" ] }, "codec": { "@AGGCAGAA": { "barcode": [ "AGGCAGAA" ] }, "@CGTACTAG": { "barcode": [ "CGTACTAG" ] }, "@GGACTCCT": { "barcode": [ "GGACTCCT" ] }, "@TAAGGCGA": { "barcode": [ "TAAGGCGA" ] }, "@TCCTGAGC": { "barcode": [ "TCCTGAGC" ] } } "algorithm": "pamld", "noise": 0.02, "confidence threshold": 0.95, } }Example 1.6 A
sampledecoder directive declaration using the phred-adjusted maximum likelihood decoder. Thetransformdirective is used to extract observed segments from the raw read while thecodecdirective names the possible barcode sequences we expect to find.
In this example we declare a sample directive that uses the phred-adjusted maximum likelihood decoder algorithm. This algorithm will choose a barcode using a maximum likelihood estimate and reject any classification with a decoding confidence lower than the confidence threshold parameter. The noise parameter is the prior probability that an observed sequence has not originated from any of the provided barcodes. The value of noise is often set to the amount of PhiX Control Library spiked into the solution for reads sequenced on the Illumina platform but can be higher if you expect other types of noise to be present. In the codec directive we provide a discrete set of possible decoding results. All barcode segment arrays must match the layout declared in the embedded transform directive, in this example one 8bp segment. The keys of the codec dictionary can be any unique string.
{ "input": [ "000000000-BDGGG_S1_L001_R1_001.fastq.gz", "000000000-BDGGG_S1_L001_I1_001.fastq.gz", "000000000-BDGGG_S1_L001_R2_001.fastq.gz" ], "template": { "transform": { "token": [ "0::", "2::" ] } }, "sample": { "transform": { "token": [ "1::8" ] }, "codec": { "@AGGCAGAA": { "barcode": [ "AGGCAGAA" ], "LB":"trinidad 5" }, "@CGTACTAG": { "barcode": [ "CGTACTAG" ], "LB":"trinidad 4" }, "@GGACTCCT": { "barcode": [ "GGACTCCT" ], "LB":"trinidad 9" }, "@TAAGGCGA": { "barcode": [ "TAAGGCGA" ], "LB":"trinidad 1" }, "@TCCTGAGC": { "barcode": [ "TCCTGAGC" ], "LB":"trinidad 8" } } "algorithm": "pamld", "noise": 0.02, "confidence threshold": 0.95, }, "CN": "CGSB", "DT": "2018-02-25T07:00:00+00:00", "PI": "300", "PL": "ILLUMINA", "PM": "miseq", "SM": "trinidad", "flowcell id": "000000000-BDGGG", "flowcell lane number": 1 }Example 1.7 A complete instruction for demultiplexing with one 8bp barcode segment present on the second input segment to an interleaved SAM stream. The CN, DT, PI, PL, PM and SM tags are declared globally and will be added to all read groups, while the LB is declared individually for each read group.
Sample barcodes are traditionally mapped to the SAM concept of Read groups. In addition to the correct sequence identifying the read group, the SAM RG header tag can contain additional metadata fields that you can either specify for an individual read group or globally for inclusion in all read groups.
@HD VN:1.0 SO:unknown GO:query @RG ID:000000000-BDGGG:1:AGGCAGAA CN:CGSB DT:2018-02-25T07:00:00+00:00 LB:trinidad 5 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:AGGCAGAA SM:trinidad @RG ID:000000000-BDGGG:1:CGTACTAG CN:CGSB DT:2018-02-25T07:00:00+00:00 LB:trinidad 4 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:CGTACTAG SM:trinidad @RG ID:000000000-BDGGG:1:GGACTCCT CN:CGSB DT:2018-02-25T07:00:00+00:00 LB:trinidad 9 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:GGACTCCT SM:trinidad @RG ID:000000000-BDGGG:1:TAAGGCGA CN:CGSB DT:2018-02-25T07:00:00+00:00 LB:trinidad 1 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:TAAGGCGA SM:trinidad @RG ID:000000000-BDGGG:1:TCCTGAGC CN:CGSB DT:2018-02-25T07:00:00+00:00 LB:trinidad 8 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:TCCTGAGC SM:trinidad @RG ID:000000000-BDGGG:1:undetermined CN:CGSB DT:2018-02-25T07:00:00+00:00 PI:300 PL:ILLUMINA PM:miseq PU:000000000-BDGGG:1:undetermined SM:trinidad M02455:162:000000000-BDGGG:1:1101:10000:10630 77 * 0 0 * * 0 0 CTAAGAAATAGACCTAGCAGCTAAAAGAGGGTATCCTGAGCCTGTCTCTTA CCCCCGGGFGGGAFDFGFGGFGFGFGGGGGGGDEFDFFGGFEFGCFEFGEG RG:Z:000000000-BDGGG:1:GGACTCCT BC:Z:GGACTCCT QT:Z:B@CCCFC< XB:f:1.56496e-06 M02455:162:000000000-BDGGG:1:1101:10000:10630 141 * 0 0 * * 0 0 GCTCAGGATACCCTCTTTTAGCTGCTAGGTCTATTTCTTAGCTGTCTCTTA CCCCCGGGGGGGGGGGGGGGGGGGF<FGGGGGGGGGGGGFGFGGGGGGGGG RG:Z:000000000-BDGGG:1:GGACTCCT BC:Z:GGACTCCT QT:Z:B@CCCFC< XB:f:1.56496e-06 M02455:162:000000000-BDGGG:1:1101:10000:12232 77 * 0 0 * * 0 0 GTATAGGGGTCACATATAGTTGGTGTGCTTTGTGAACTGCGATCTTGACGG CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG RG:Z:000000000-BDGGG:1:GGACTCCT BC:Z:GGACTCCT QT:Z:CCCCCGGG XB:f:1.56086e-06 M02455:162:000000000-BDGGG:1:1101:10000:12232 141 * 0 0 * * 0 0 GTCCTATCCTACTCGGCTTCTCCCCATTTTTCAGACATTTTCCTATCAGTC CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG RG:Z:000000000-BDGGG:1:GGACTCCT BC:Z:GGACTCCT QT:Z:CCCCCGGG XB:f:1.56086e-06Example 1.8 Output header and first 4 records (two complete reads) from demultiplexing using the configuration file in Example 1.7. Notice how tags declared globally were added to all read groups. The RG and PU read group identifiers default to the convention set by GATK if you provide the flowcell related directives. The XB tag reports the probability the read was incorrectly classified.
Providing Barcode Priors
The PAML decoder computes the posterior probability that the decision it made was correct for each classified read, that probability depends on a set of priors. If the 5 libraries were pooled in non uniform concentrations we will expect the portion of reads classified to each read group to match those proportions. A prior on the barcode prevalence distribution can be provided for each possible code declared in the codec directive using the concentration parameter. For convenience the priors do not have to be specified as normalized probabilities. Pheniqs will normalize them when compiling the instructions to sum up to 1.0 minus the value of the noise parameter.
{ "sample": { "transform": { "token": [ "1::8" ] }, "codec": { "@AGGCAGAA": { "barcode": [ "AGGCAGAA" ], "concentration": 2 }, "@CGTACTAG": { "barcode": [ "CGTACTAG" ] }, "@GGACTCCT": { "barcode": [ "GGACTCCT" ] }, "@TAAGGCGA": { "barcode": [ "TAAGGCGA" ] }, "@TCCTGAGC": { "barcode": [ "TCCTGAGC" ] } } "algorithm": "pamld", "noise": 0.02, "confidence threshold": 0.95, } }Example 1.9 Adding a prior to one of the code words when declaring a
sampledecoder directive. Since the priors are automatically normalized and default to 1, this declaration effectively states that we expect twice as many reads to be classified to @AGGCAGAA than the other 4 read groups.
Minimum Distance Decoding
Pheniqs can also be instructed to decode the barcodes using the traditional minimum distance decoder, that only consults the edit distance between the expected and observed sequence, by setting the sample decoder algorithm directive to mdd. The MDD decoder however ignores the presence of noise and the error probabilities provided by the sequencing instrument and does not compute or report the classification error probability. It is provided for legacy purposes but PAMLD will yield superior results in almost every real world scenario.
Speeding things up
As we mentioned before reading input from CRAM or BAM input can be faster than reading from gzip compressed FASTQ files. If your first step was to package the 3 split FASTQ files into a CRAM file, as shown in Example 1.3, you can use that file as input.
{ "input": [ "000000000-BDGGG_raw.cram", "000000000-BDGGG_raw.cram", "000000000-BDGGG_raw.cram" ], "template": { "transform": { "token": [ "0::", "2::" ] } }, "sample": { "transform": { "token": [ "1::8" ] }, "codec": { "@AGGCAGAA": { "barcode": [ "AGGCAGAA" ] }, "@CGTACTAG": { "barcode": [ "CGTACTAG" ] }, "@GGACTCCT": { "barcode": [ "GGACTCCT" ] }, "@TAAGGCGA": { "barcode": [ "TAAGGCGA" ] }, "@TCCTGAGC": { "barcode": [ "TCCTGAGC" ] } } "algorithm": "pamld", "noise": 0.02, "confidence threshold": 0.95, }, "CN": "CGSB", "DT": "2018-02-25T07:00:00+00:00", "PI": "300", "PL": "ILLUMINA", "PM": "miseq", "SM": "trinidad", "flowcell id": "000000000-BDGGG", "flowcell lane number": 1 }Example 1.10 modifying Example 1.7 to take the CRAM file created in Example 1.3 as input. When declaring interleaved input you specify the file path as many times as the interleaving resolution. The interleaving resolution is the number of consecutive segments of the same read that have been interleaved into the file. In this example we expect every read to have 3 consecutive segments.
The output from Example 1.10 will be identical to the output from Example 1.7 shown in Example 1.8 but unlike Example 1.7 the decoding speed will scale linearly with the number of computational cores available until the system’s I/O throughput becomes saturated.