Read segment transformation in Pheniqs
At the heart of Pheniqs is a transformation framework that relies on a familiar syntax that mimics Python array slicing and can arbitrarily manipulate read structure and decode multiple barcodes anywhere in a sequence read. It extracts tokens from multiple read segments by addressing either the 5’ end, 3’ end, or both (and optionally reverse complement) to construct the output template segments and the sample, cellular and molecular barcodes. This generic approach can accommodates any potential barcoding scheme and obviates the need for pre and post processing for most experimental designs.
Experimental Design
Some very common designs for Illumina platforms are illustrated in the following diagram.

More complicated barcoding schemes for single-cell, CRISPR, and multi-modal sequencing are also appearing. Examples of how to configure Pheniqs for a variety of experimental designs, may be found in the vignettes section of the documentation. Template configuration files with common barcode sets are also available in the template section
Standard Illumina Read anatomy
Illumina sequencing platforms typically produce four different sequence elements: two Index sequences, referred by Illumina as the I5 and I7 barcodes, and two Insert sequences, referred by Illumina as Read 1 and Read 2. Collectively, these are referred to by the sequence alignment map format specification as read segments. For example, consider a standard paired-end, dual index library design:
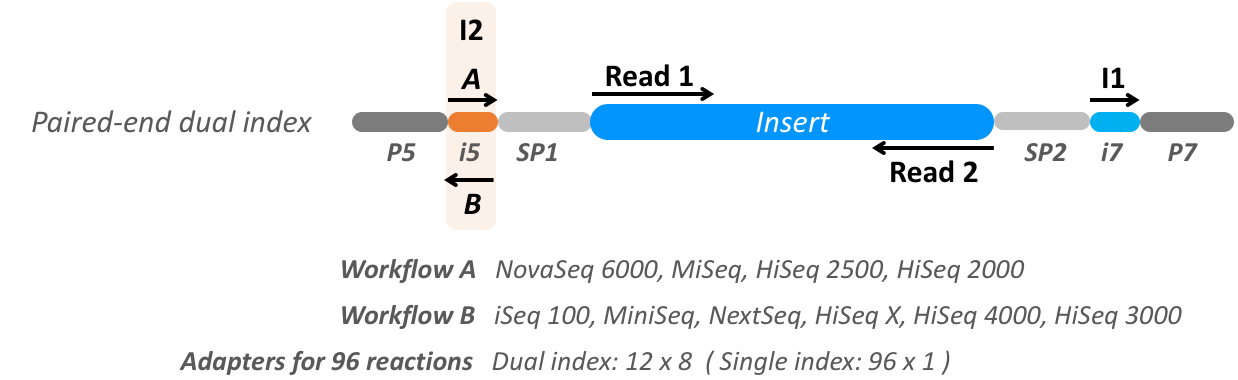
The read segments for this standard design thus comprise two technical sequences (referred by Illumina file naming terminology as I1, I2) and two biological sequences (referred by Illumina file naming terminology as R1, R2):
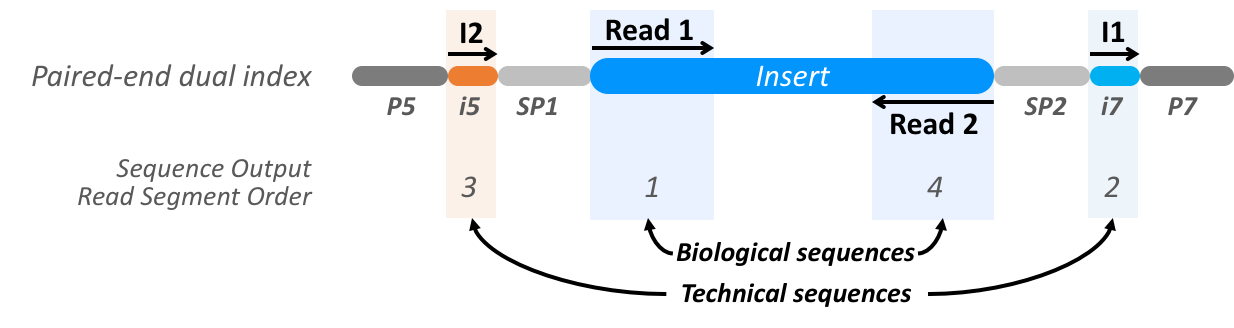
The four segments are then provided in the order: R1, I1, I2, R2. Together those for segments constitute a single read and all share the same identifier, whether in FASTQ format or SAM. Whgen using SAM format, the segment index may be indicated in one of two ways: when the read has no more than 2 segments the 0x40 and 0x80 bits of the SAM flag mark the first segment in the template and the last segment in the template. When more than 2 segments are present the FI auxiliary tag provides the index of segment in the template and the TC auxiliary tag provides the total number of segments in the template. Encoding of the segment index in the FASTQ format is not standardized but in reads produced by the Illumina platform will often be present in the comment section, which appears on the same line as the identifier folowing a whitespace character.
Read Classification
The combination of the barcodes contained in the I1 and I2 index segments identifies the pooled library in the standard illumina protocol. With standard dual indexing, up to 96 distinct sample libraries can be pooled and run together in a single sequencing lane.
To identify which biological sequences belong to which library, the sequences belonging each one need to be separated from each other. This process of deconvolving libraries is called demultiplexing and is done by classifying each of the sequences using the barcode indexes:
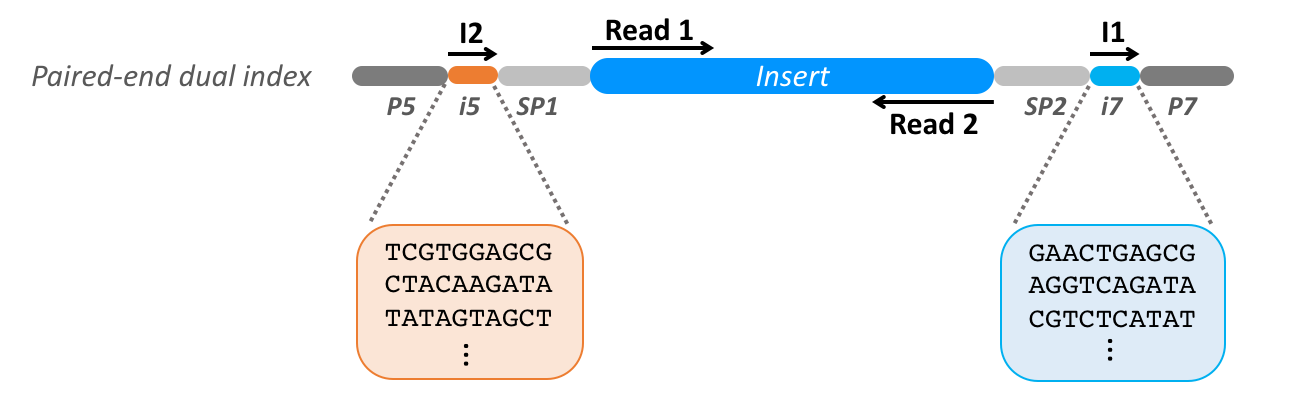
Note The i5 adaptor sequences specified in sample sheets will be reverse complemented for platforms that read the I2 index on the bottom strand. The i7 sequences in sample sheets are always reverse complemented relative to the original adaptor sequences since they are read from the top strand.
Tokenization
Pheniqs uses tokens to reference and extract information from different read segments by specifying where to look for different classes of sequence elements (i.e. barcodes, biological sequences). Each element of interest is defined by an offset relative to the beginning of a given read segment (in this example I1, I2, R1, R2) and an end coordinate. It is important to note that Pheniqs uses zero based indexing, so the first read to come off the machine will be referred to as Segment 0, and so on:
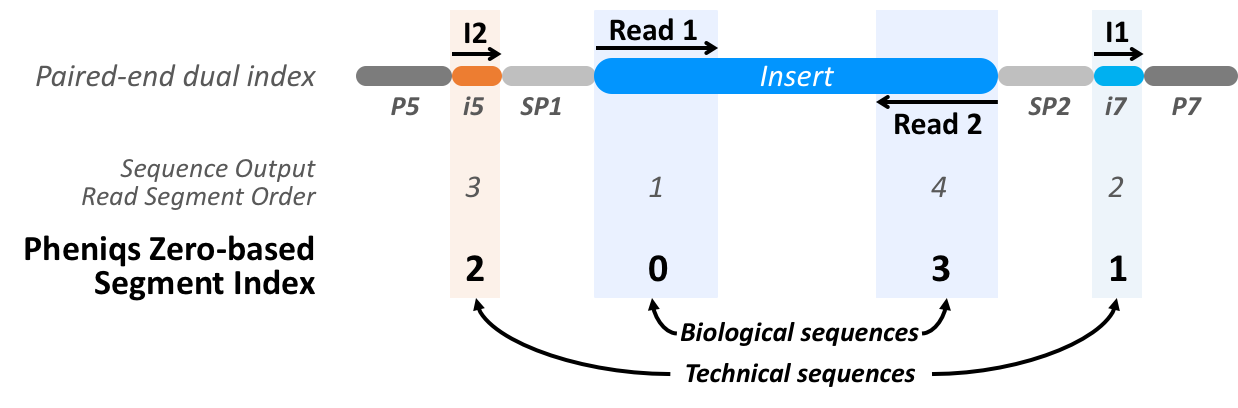
For this design, the barcode tokens begin at position 0 in I1 and I2 and extend for 10 bases. The tokens for biological sequences begin at position 0 of Read 1 and Read 2 and extend for the full span of those read segments.
For a standard paired-end, dual indexed Illumina run, the sample barcodes usually comprise the full I1 and I2 read segments. Because Illumina sequencing is calibrated in relation to the previously sequenced base, sequencing centers sometimes sequence those segments one nucleotide longer than necessary to ensure good quality on the last nucleotide, so it is a good idea to explicitly provide an end coordinate when tokenizing the index segments. The biological sequences start at the first position of R1 and R2 and extend for the full number of cycles run (typically 75, 100, or 150 nucleotides).
Transform Patterns
This example illustrates tokenization syntax and output for a 150nt dual-indexed paired-end sequencing run with sample, cellular, and molecular barcodes. This example contains the following features:
- A sample barcode composed of two 10nt elements (i5 and i7)
- A 12nt inline cellular barcode (Cell)
- A 12nt inline molecular barcode (UMI)
- An Insert containing the biological sequence of interest (template), which is sequenced from both ends. The template sequences are located in Read 1 (31nt just downstream of the Cell and UMI) and all of Read 2 (here, 75nt).
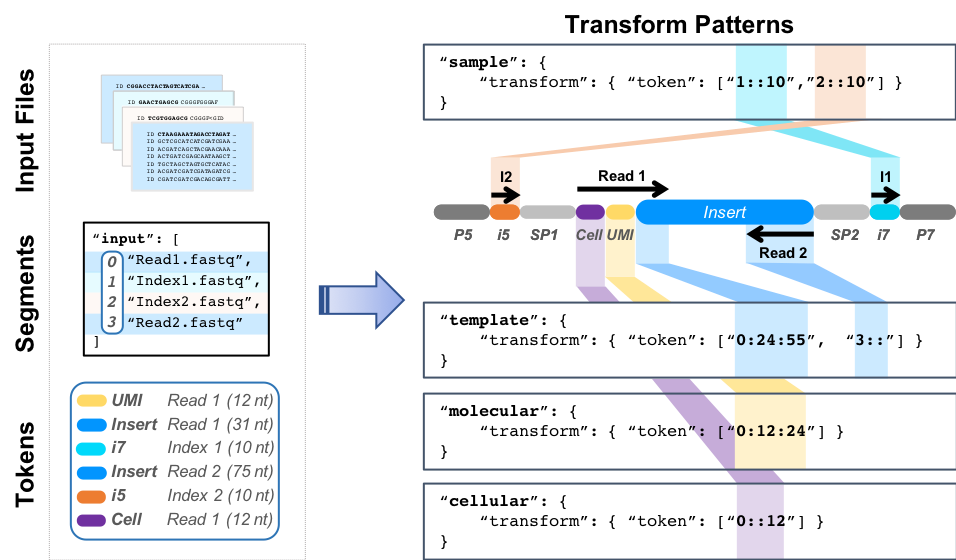
Input files contain sequence data to be analyzed. For Illumina data, these will usually be in split fastq format and there will be one file for each read segment that comes off the sequencing machine. The read segments are catalogued for future reference and are indexed as an array, where 0=Read1, 1=Index1, 2=Index2, 3=Read2. Barcode tokens define the type and location of the sequences of interest in each read segment. Barcodes may appear at any position and orientation in any read segment. Each type of barcode included in the experimental design is designated as a separate entity. The transform patterns define how each sequence component to be extracted is to be handled. Each token comprises three colon separated components,
segment:start:end. Per Python array slicing syntax, the start coordinate (offset) is inclusive and the end coordinate is exclusive. Start and end coordinates default to 0 and the end of the segment, respectively.
Output
After sequence classification, the tokenized sequences that have been extracted are written along with their associated metadata, including sequence quality and confidence scores, to specific auxiliary tags within the SAM format. While Pheniqs can also produce FASTQ output, SAM is preferred since it preserves all associated metadata.

Sample output for the 150nt paired-end dual-indexed experimental design shown above. Template read segments are emitted along with observed and most likely inferred barcode sequences, quality scores, and error probabilities. The confidence score for each token is one minus its estimated error based on the full posterior probability of observation; for the compound sample barcode here, it is the product of the confidence scores for each component and is one minus the error probability shown.
The SAM tags populated by Pheniqs are summarized below:
| Name | Description | Example |
|---|---|---|
| RG | Read group identifier matching an RG:ID field in the header. | H7LT2DSXX:1:GAACTGAGCGTCGTGGAGCG |
| BC | Raw uncorrected sample barcode sequence. | GAACTGAGCG-TCGTGGAGCG |
| QT | Phred quality of the sample barcode sequence in the BC tag. | ,FF::F:F:F-,,FF::FF:F |
| XB | The probability that sample barcode decoding is incorrect. | 2.27479e-06 |
| CB | Cellular identifier. | ACTGCATA |
| CR | Raw uncorrected cellular barcode sequence. | ACTGCATT |
| CY | Phred quality of the cellular barcode sequence in the CR tag. | ,,FF::FF |
| XC | The probability that Cellular barcode decoding is incorrect. | 2.27479e-06 |
| MI | Molecular Identifier. | |
| RX | Molecular barcode sequence, either corrected or uncorrected. | |
| QX | Phred quality of the molecular barcode sequence in the RX tag. | |
| OX | Raw uncorrected molecular barcode sequence. | |
| BZ | Phred quality of the molecular barcode sequence in the OX tag. | |
| XM | The probability that molecular barcode decoding is incorrect. |