Standard Illumina sample decoding with the python API
This tutorial will walk you through prior estimation and demultiplexing of a standard Illumia sequencing run using pheniqs-illumina-api.py, pheniqs-prior-api.py and pheniqs-io-api.py. In this example we use the python API to generate configuration files for PAMLD from metadata found in an output Illumina run folder for flowcell H7LT2DSXX. For an overview of manually write such configuration files for a similar scenario see the Standard Illumina sample decoding. This example also demonstrates using the import directive to cascade configuration files.
Sample barcode sequence and quality scores will be written to the BC and QT auxiliary tags. The decoding error probability will be stored in the XB tag.
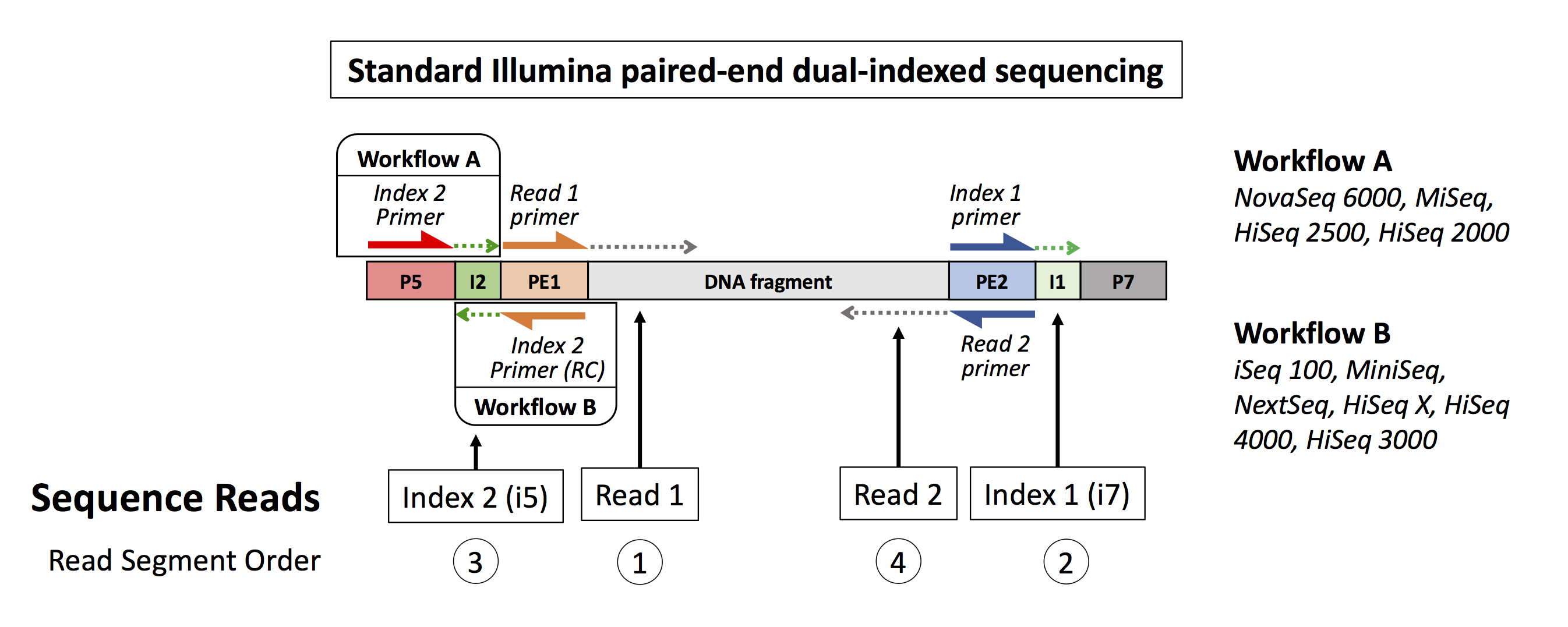
The run has paired end dual index samples multiplexed using the standard Illumina i5 and i7 index protocol. Each read has 4 segments: two 151 base pairs long biological sequences from the DNA or RNA fragment and two 8 base pairs long technical sequences containing the i5 and i7 indices.
All shell commands bellow are executed in the H7LT2DSXX example directory, where you can also find all other files related to this example.
Base calling
Base calling is done with the Illumina bcl2fastq utility. Since we will want to decode barcodes by other means we need to adjust bcl2fastq’s configuration to simply write the 4 segments of the reads to 4 files. The basecall sub command of pheniqs-illumina-api.py can generate a bcl2fastq shell command for base calling without decoding the sample barcodes.
pheniqs-illumina-api.py basecall \ --output-dir . \ --fastq-compression-level 3 \ 181014_A00534_0024_AH7LT2DSXXgenerating a bcl2fastq command we want output fastq files to be written to this directory and we want lower gzip compression since we will not be archiving those files.
This will produce H7LT2DSXX_basecall.sh and H7LT2DSXX_basecall_sample_sheet.csv. pheniqs-illumina-api.py basecall allows you to forward several relevant bcl2fastq parameters to be included in the script, but you can also manually adjust it.
bcl2fastq \ --runfolder-dir 181014_A00534_0024_AH7LT2DSXX \ --sample-sheet H7LT2DSXX_basecall_sample_sheet.csv \ --create-fastq-for-index-reads \ --adapter-stringency 0 \ --minimum-trimmed-read-length 0 \ --mask-short-adapter-reads 0 \ --output-dir . \ --fastq-compression-level 3bcl2fastq shell command We must provide bcl2fastq an alternative sample sheet or it will default to using the one in the run folder. A simple sample sheet that does not perform any barcode decoding is generated by
pheniqs-illumina-api.py basecall. Base calling with bcl2fastq took about 6 hours on a dual socket Intel Xeon E5-2620 with a total of 12 cores and produced 4 files per lane.
H7LT2DSXX_S1_L001_I1_001.fastq.gz H7LT2DSXX_S1_L001_I2_001.fastq.gz H7LT2DSXX_S1_L001_R1_001.fastq.gz H7LT2DSXX_S1_L001_R2_001.fastq.gz H7LT2DSXX_S1_L002_I1_001.fastq.gz H7LT2DSXX_S1_L002_I2_001.fastq.gz H7LT2DSXX_S1_L002_R1_001.fastq.gz H7LT2DSXX_S1_L002_R2_001.fastq.gz H7LT2DSXX_S1_L003_I1_001.fastq.gz H7LT2DSXX_S1_L003_I2_001.fastq.gz H7LT2DSXX_S1_L003_R1_001.fastq.gz H7LT2DSXX_S1_L003_R2_001.fastq.gzbcl2fastq output 2 biological and 2 technical sequences were produced by bcl2fastq base calling.
R1containing the 3 prime prefix of the insert region,I1containing the i7 index,I2containing the i5 index andR2containing the reverse complemented 5 prime suffix of the insert region, since it was read in reverse.
Core configuration
H7LT2DSXX_core.json summarizes metadata extracted from RunInfo.xml and SampleSheet.csv. It is not meant to be executed by itself but is imported by most other configuration files in this tutorial. To generate one use the core subcommand of pheniqs-illumina-api.py
pheniqs-illumina-api.py core \ --base-input . \ --base-output . \ --no-input-npf \ 181014_A00534_0024_AH7LT2DSXXGenerating a core configuration
--no-input-npfwill add a globalfilter incoming qc failinstruction to discard reads that failed the internal Illumina sequencer chastity filter from the incoming feed. Any parameter we provide on the core configuration will percolate to every configuration file that imports it, so its a good place to provide global metadata.
H7LT2DSXX_core.json conveniently declares an array of decoders that where recovered from the SampleSheet.csv file as well as transformation rules from RunInfo.xml. When you import H7LT2DSXX_core.json, you can reuse those in your configuration file and expand them so you don’t need to constantly be editing big configuration files. Anything specified in a configuration file will override an imported directive.
{ "PL": "ILLUMINA", "PM": "A00534", "flowcell id": "H7LT2DSXX" }core configuration already declares PL, PM, and
flowcell id. Those were extracted from RunInfo.xml but you can modify them.
decoder configuration element
H7LT2DSXX_core.json declares a decoder directive, a dictionary of abstract decoders.
{ "decoder": { "H7LT2DSXX_l01_multiplex": { "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "barcode": [ "CGAGGCTG", "GTAAGGAG" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "barcode": [ "AAGAGGCA", "ACTGCATA" ] } }, "transform": { "token": [ "1::8", "2::8" ] } } } }H7LT2DSXX_l01_multiplex decoder lists the possible barcode combinations and the library names associated with them in the LB tag extracted from SampleSheet.csv.
In the importing file, you can use the base property in a decoder to use those as a starting point in the sample, cellular, and molecular. Any directive you specify in your instantiation will override values provided by the referenced base. You will see an example of how it is used in the next section.
Sample barcode decoding
The sample sub command will generate a basic sample demultiplexing configuration file for each lane.
pheniqs-illumina-api.py sample \ --confidence 0.95 \ --noise 0.05 \ 181014_A00534_0024_AH7LT2DSXXpheniqs-illumina-api sample will produce a basic sample demultiplexing configuration that we will adjust later with the prior estimates. Some pheniqs parameters can be forwarded like
--noiseand--confidenceshown in this example.
H7LT2DSXX_l01_sample.json
is a configuration for demuliplexing the first lane. It declares a sample directive that expands the H7LT2DSXX_l01_multiplex decoder we have seen defined in H7LT2DSXX_core.json.
{ "import": [ "H7LT2DSXX_core.json" ], "input": [ "H7LT2DSXX_S1_L001_R1_001.fastq.gz", "H7LT2DSXX_S1_L001_I1_001.fastq.gz", "H7LT2DSXX_S1_L001_I2_001.fastq.gz", "H7LT2DSXX_S1_L001_R2_001.fastq.gz" ], "sample": { "algorithm": "pamld", "base": "H7LT2DSXX_l01_multiplex", "confidence threshold": 0.95, "noise": 0.05 }, "output": [ "H7LT2DSXX_l01.bam" ], "report url": "H7LT2DSXX_l01_sample_report.json" }Configuration for decoding with a uniform prior in this case, the output is interleaved into a single bam file.
Estimating noise and sample priors
To estimate priors for sample barcodes we need to collect some statistics. The estimate sub command will generate, for each lane, an optimized configuration for sample prior estimation.
pheniqs-illumina-api.py estimate \ --confidence 0.95 \ --noise 0.05 \ 181014_A00534_0024_AH7LT2DSXXpheniqs-illumina-api estimate will produce a sample demultiplexing configuration optimized for prior estimation.
Since it is not necessary to read the biological segments when estimating the priors, input is declared only for the two index segments.
H7LT2DSXX_l01_estimate.json declares a sample directive that expands H7LT2DSXX_l01_multiplex and adjusts the tokenization for the modified input. output is redirected to /dev/null to tell Pheniqs it should not bother with the output.
{ "import": [ "H7LT2DSXX_core.json" ], "input": [ "H7LT2DSXX_S1_L001_I1_001.fastq.gz", "H7LT2DSXX_S1_L001_I2_001.fastq.gz" ], "sample": { "algorithm": "pamld", "base": "H7LT2DSXX_l01_multiplex", "confidence threshold": 0.95, "noise": 0.05, "transform": { "token": [ "0::", "1::" ] } }, "output": [ "/dev/null" ], "report url": "H7LT2DSXX_l01_estimate_report.json", "template": { "transform": { "token": [ "0::", "1::" ] } } }Prior estimation configuration refrains from reading the biological sequences and produces no output which significantly speeds things up..
Executing this configuration will yield the H7LT2DSXX_l01_estimate_report.json report. Like every Pheniqs report, it contains decoding statistics that we can use to estimate the priors.
pheniqs mux --config H7LT2DSXX_l01_estimate.jsonexecuting pheniqs with a prior estimation configuration will produce H7LT2DSXX_l01_estimate_report.json. This took about 1:45 hours per lane on our dual socket Intel Xeon E5-2620.
Now that you have H7LT2DSXX_l01_sample.json, a sample decoding configuration, and H7LT2DSXX_l01_estimate_report.json, a report with decoding statistics, you can use pheniqs-prior-api.py to generate a prior adjusted configuration file.
pheniqs-prior-api.py \ --report H7LT2DSXX_l01_estimate_report.json \ --configuration H7LT2DSXX_l01_sample.json \ H7LT2DSXX_l01_adjusted.jsondecoding a lane with an estimated prior H7LT2DSXX_l01_adjusted.json is similar to H7LT2DSXX_l01_sample.json with the addition of the estimated priors.
IO manipulation
If you need the output to be split into files by either library or segment you can use pheniqs-io-api.py to adjust the necessary directives in your configuration. Splitting by segment means every read segment is written to a separate file, rather than interleaved into the same file. Splitting by library means reads classified to different libraries are written to different files.
pheniqs-io-api.py \ --configuration H7LT2DSXX_l01_adjusted.json \ --format fastq \ --split-segment \ --split-library \ H7LT2DSXX_l01_adjusted_split.jsonsplitting both library and segment will adjust the output directives in the configuration file and generate the necessary file names for splitting by both library and segment.
{ "sample": { "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "output": [ "H7LT2DSXX_A10_PDAC81_s01.fastq.gz", "H7LT2DSXX_A10_PDAC81_s02.fastq.gz" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "output": [ "H7LT2DSXX_A11_PDAC490_s01.fastq.gz", "H7LT2DSXX_A11_PDAC490_s02.fastq.gz" ] } }, "undetermined": { "output": [ "H7LT2DSXX_undetermined_s01.fastq.gz", "H7LT2DSXX_undetermined_s02.fastq.gz" ] } } }Configuration adjustments for splitting by library and segment adjustments made by
pheniqs-io-api.py. Global output directives are stripped and each barcode is generated a new output directive. In this case we split by both segment and library and output gzip compressed fastq.
pheniqs-io-api.py \ --configuration H7LT2DSXX_l01_adjusted.json \ --format bam \ --split-library \ H7LT2DSXX_l01_adjusted_split.jsonsplitting by library when working with SAM encoded files it makes less sense to split the segments into separate files.
{ "sample": { "codec": { "@A10_PDAC81": { "LB": "A10_PDAC81", "output": [ "H7LT2DSXX_A10_PDAC81.bam" ] }, "@A11_PDAC490": { "LB": "A11_PDAC490", "output": [ "H7LT2DSXX_A11_PDAC490.bam" ] } }, "undetermined": { "output": [ "H7LT2DSXX_undetermined.bam" ] } } }Configuration adjustments for splitting by library adjustments made by
pheniqs-io-api.py. Global output directives are stripped and each barcode is generated a new output directive. In this case we split by library and interleave all segments into bam files.
Decoding with the estimated prior
Now that you have a prior adjusted configuration file you can execute it with Pheniqs.
pheniqs mux --config H7LT2DSXX_l01_adjusted.jsondecoding a lane with an estimated prior the report will be written to H7LT2DSXX_l01_sample_report.json because it is relative to the base output. Decoding each of the 4 lanes took about 5:40 hours and produced a 488GB bam file on our dual socket Intel Xeon E5-2620.