Pheniqs
Pheniqs is a flexible generic barcode classifier for high-throughput next-gen sequencing that caters to a wide variety of experimental designs and has been designed for efficient data processing. Features include:
Citing Pheniqs: Pheniqs 2.0: accurate, high-performance Bayesian decoding and confidence estimation for combinatorial barcode indexing on BMC Bioinformatics.
Please check out the intro talk given by Lior Galanti on April 29, 2021 for the NYU CGSB core lab.
Powerful and intuitive syntax
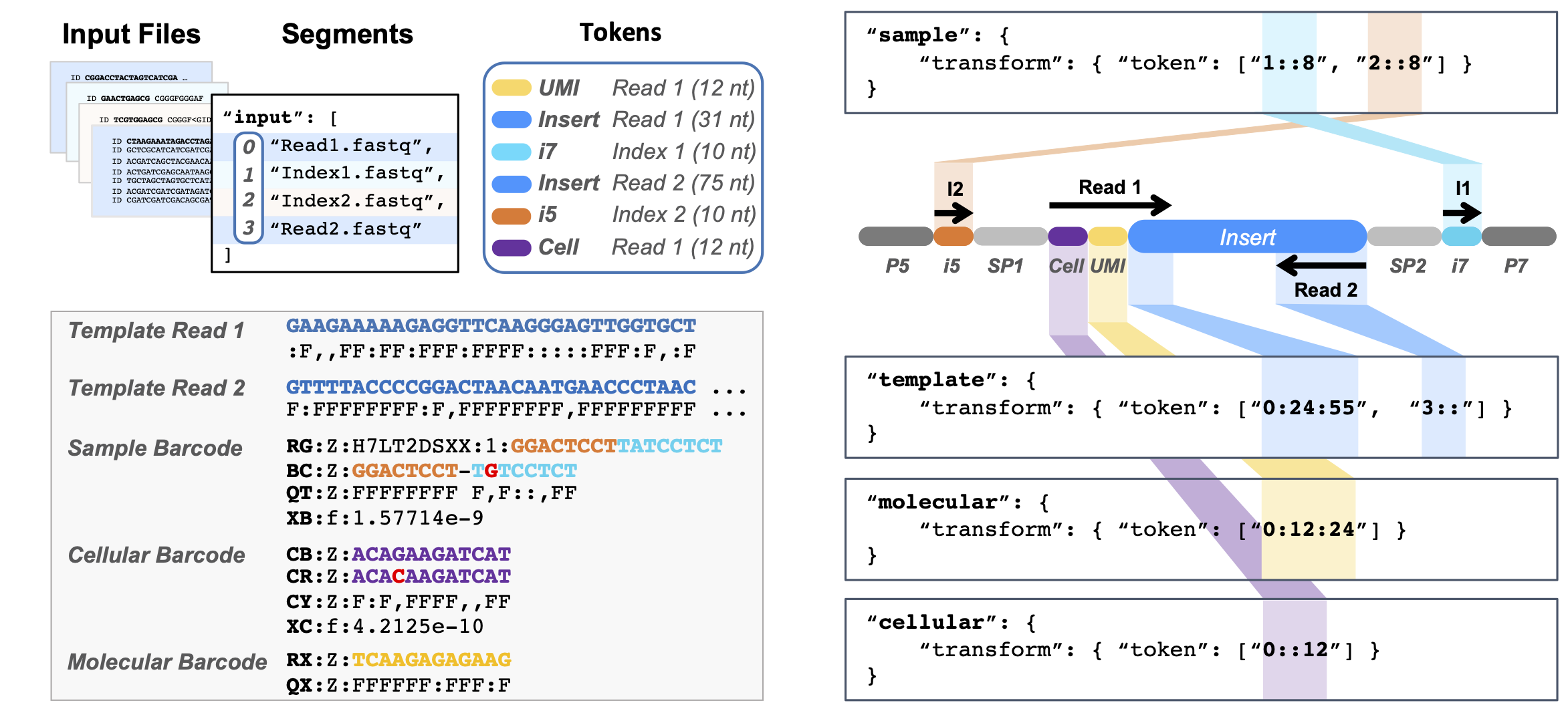
- Classifies standard barcode types: Sample, Cellular, and Molecular Index
- Directly writes barcodes to standard or custom BAM fields
- Addresses index tags in arbitrary locations along reads
- Easily accommodates custom barcode types, eliminating the need for pre- or post-processing
- Easily handles any number of combinatorial barcode tags
Noise and quality aware probabilistic classifier
- Increased accuracy over standard edit distance methods
- Reports classification error probabilities in SAM auxiliary tags
- Modular design allows addition of new classifiers
Robust engineering
- Multithreaded C++ implementation optimized for speed
- POSIX standard stream integration
- Directly interfaces with low level HTSLib C API
- Performance scales linearly with the number of available processing cores
Easy to install or build
- Stable releases available from Bioconda
- Custom package manager can build dependencies and binaries from scratch
- Easily installed on clusters or cloud without elevated permissions
- Portable compiled binaries available
- Available in a Docker container
Easy to use
- Simple command line syntax with autocomplete
- Reusable, inheritence enabled, JSON encoded configuration
- Preconfigured barcode library sets
- Reads and writes multiple file formats: FASTQ, SAM/BAM/CRAM
- Fast standalone file format interconversion
- Helper scripts to assist in configuration file bootstraping
- Facilitates more robust and reproducible downstream analysis
On this website, you will find everything you need to get started with Pheniqs: Installation and configuration, classifying reads, vignettes that will walk you through processing popular experimental designs, and information about how to leverage standardized SAM auxiliary tags to improve the reproducibility of your published data.
Pheniqs runs on all modern POSIX systems and provides an easy to learn command line interface with autocomplete and an extensible reusable configuration syntax. Pheniqs is an ideal utility to pre- and post-process sequence reads for other bioinformatics tools, and it may also be used simply to rapidly and efficiently interconvert a variety of standard sequence file formats without invoking any of its barcode processing features.
For more advanced users and sequencing core managers, we provide detailed build instructions and a custom package manager to easily build portable, statically linked, Pheniqs binaries for deployment on computing clusters. Developers can find code examples and API documention that enable them to expand Pheniqs with new classification algorithms and take advantage of the optimized multithreaded pipeline.
Installing Pheniqs
You can install pheniqs using the conda package manager on Linux
conda install -c bioconda -c conda-forge pheniqs
You can build the latest Pheniqs binary with this one line. Just paste it into a Linux or macOS terminal.
bash -c "$(curl -fsSL https://raw.githubusercontent.com/biosails/pheniqs-build-api/master/install_pheniqs.sh)"