- Configuration syntax
- Format support and performance considerations
- The
inputdirective - The
transformdirective - Barcode decoding
- URL handling
- Phred offset
- Leading Segment
- Pass filter / QC fail reads
- Configuration validation and compliation
- Quality Control and Statistics
Configuration syntax
When executing trivial scenarios, such as format conversion or interleaving, it is sufficient to provide parameters on the command line. However, scenarios involving barcode decoding are too verbose to provide on a command line and require a JSON encoded configuration file containing directives for input and output layout, read segment manipulation, barcode decoding and other run parameters. Parameters specified as command line arguments always override their configuration file counterparts. Almost all parameters have a default value. A brief description of the parameters available on the command line is available with the -h/--help flags. If you use zsh the bundled command line completion will give you a more interactive command line experience.
A Pheniqs configuration file can import instructions from additional configuration files and supports a sophisticated inheritance mechanism. While you may certainly ignore this added complexity at first, separating different aspects of your experimental design into reusable instruction files can significantly simplify day to day use. Standard Illumina codecs are also available as generic configuration files you can import and inherit from.
Format support and performance considerations
Pheniqs can arbitrarily manipulate reads stored in either the advanced HTS containers SAM, BAM and CRAM or the FASTQ format with segments either interleaved into a single file or split over many. Some features are only supported with HTS containers, since FASTQ records have no standardized method of associating metadata with read segments. Pheniqs is written in highly optimized multi threaded C++11 and interfaces directly with the low level HTSlib API. It can be installed from many popular package managers or compiled from source into a single portable executable binary using the pheniqs build tool.
Pheniqs execution speed will normally scale linearly with the number of cores available for computation. However since the gzip compression algorithm does not scale well in multi threaded environments, reading or writing from gzip compressed FASTQ files on machines with many cores will often be I/O bound. You can use Pheniqs to easily repack data stored in FASTQ files into an efficient, interleaved, CRAM or BAM container. CRAM and BAM offers significant improvements in both performance and storage requirements as well as support for associating extensible metadata with read segments.
Pheniqs aims to fill the gap between advanced HTS containers and analysis tools that only supports FASTQ. By efficiently converting between HTS and FASTQ over standard streams, it allows you to feed analysis software with FASTQ directly from annotated HTS files, without additional storage requirements.
The input directive
The input directive is an ordered array of file paths. Pheniqs assembles an input read by reading one segment from each input file. The number of file paths in the input array determines how many segments an input read has.
{ "input": [ "HK5NHBGXX_S1_L001_R1_001.fastq.gz", "HK5NHBGXX_S1_L001_I1_001.fastq.gz", "HK5NHBGXX_S1_L001_I2_001.fastq.gz", "HK5NHBGXX_S1_L001_R2_001.fastq.gz", ] }Example 2.1 Declaring an input read that is split over four gzip compressed FASTQ files.
Interleaved files contain multiple consecutive segments of the same read. You can ask Pheniqs to automatically detect the interleaving pattern or explicitly assemble an input read from interleaved files by simply repeating the path to reference the same file multiple times, once for each segment.
{ "input": [ "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram" ] }Example 2.2 Declaring a 4 segment input read interleaved into a single CRAM file.
You can even mix-and-match the two layout styles
{ "input": [ "HK5NHBGXX_l01_biological.fastq.gz", "HK5NHBGXX_l01_technical.fastq.gz", "HK5NHBGXX_l01_technical.fastq.gz", "HK5NHBGXX_l01_biological.fastq.gz", ] }Example 2.3 Constructing a four segment input read from a mixed style layout. The two biological segments, 0 and 3, are interleaved in
HK5NHBGXX_l01_biological.fastq.gzwhile the two technical segments, 1 and 2, are interleaved intoHK5NHBGXX_l01_technical.fastq.gz.
The input directive defaults to expecting interleaved input from standard input. While input format is automatically detected interleaving layout is not unless you specify the -s/--sense-input command line flag.
{ "input": [ "/dev/stdin" ] }Example 2.4 This is the default input directive. If not instructed otherwise, Pheniqs implicitly expects input from standard input. The format will be automatically detected.
Automatic input sensing
If the -s/--sense-input command line flag is specified, Pheniqs will automatically detect the format of the input you provide it by examining the first few bytes. Once the input format is established, Pheniqs decodes the first few segment records from the feed to detect the feed’s resolution: the number of consecutive segments in the feed that have the same read identifier. Pheniqs will assume that to assemble a read it will have to read that many segments from each input feed.
@M02455:162:000000000-BDGGG:1:1101:10000:10630 1:N:0: CTAAGAAATAGACCTAGCAGCTAAAAGAGGGTATCCTGAGCCTGTCTCTTA + CCCCCGGGFGGGAFDFGFGGFGFGFGGGGGGGDEFDFFGGFEFGCFEFGEG @M02455:162:000000000-BDGGG:1:1101:10000:10630 2:N:0: GGACTCCT + B@CCCFC< @M02455:162:000000000-BDGGG:1:1101:10000:10630 3:N:0: GCTCAGGATACCCTCTTTTAGCTGCTAGGTCTATTTCTTAGCTGTCTCTTA + CCCCCGGGGGGGGGGGGGGGGGGGF<FGGGGGGGGGGGGFGFGGGGGGGGG @M02455:162:000000000-BDGGG:1:1101:10000:12232 1:N:0: GTATAGGGGTCACATATAGTTGGTGTGCTTTGTGAACTGCGATCTTGACGG + CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG @M02455:162:000000000-BDGGG:1:1101:10000:12232 2:N:0: GGACTCCT + CCCCCGGG @M02455:162:000000000-BDGGG:1:1101:10000:12232 3:N:0: GTCCTATCCTACTCGGCTTCTCCCCATTTTTCAGACATTTTCCTATCAGTC + CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGExample 2.5 This FASTQ file interleaves three segments for each read and so has a resolution value of 3.
The transform directive
Read manipulation is achieved with the transform directive by means of tokenization with the token array and segment assembly with the optional knit array. The same transform syntax is used in the template directive to specify how the output read is constructed, and in each decoder to construct the segmented sequence that is used to match against the dictionary of possible valid barcodes. In the tokenization step Pheniqs consults a token pattern to extract a token from an input segment. In the segment assembly step one or more segment patterns reference tokens to assemble a new segment. The optional knit directive is only necessary when assembling segments from multiple, non contiguous, tokens or if a token needs to be reverse complemented. If the knit directive is omitted from a transform directive, each token implicitly declares a single segment.
Tokenization
A token pattern is made of 3 colon separated integers. The first is the mandatory zero based input segment index enumerated by input. The second is an inclusive zero based start coordinate to the beginning of the token and defaults to 0 if omitted. The third is an exclusive zero based end coordinate to the end of the token. If the end coordinate is omitted the token spans to the end of the segment. start coordinate and end coordinate can take positive or negative values to access the segment from either the 5’ (left) or 3’ (right) end and mimic the python array slicing syntax. The two colons are always mandatory.
{ "template": { "transform": { "token": [ "0::", "1::8" ] } } }Example 2.6 In this
transformdirective the first token spans the entire first input segment. The second spans the first 8 cycles of the second segment. Since theknitdirective is omitted each token effectively declares a segment.
{ "template": { "transform": { "token": [ "0:0:", "1:0:8" ], "knit": [ "0", "1" ] } } }Example 2.7 This
transformdirective is semantically identical to the one provided in Example 2.6 but explicitly declares theknitdirective and token start coordinate.
Since token patterns follow the python array slicing syntax you can test your patterns by using a toy example of an array in a python shell. The follwing are examples of tokenizing 9 nucleotides. The Bitmap column shows you exactly which cycles are effectively selected by the pattern.
Cycle 012345678Nucleotide GGACTCCTA
Pattern Segment Start End Cycles Bitmap Description 0:0:3003GGA+++------First 3 cycles of segment 0 0:0:009GGACTCCTA+++++++++All of segment 0 0::009GGACTCCTA+++++++++All of segment 0 0:-4:059CCTA-----++++Last 4 cycles of segment 0 0:-5:-1048TCCT----++++-Cycles 1 to 5 from the 3’ end 0:3:-2037CTCC---++++--All but the first 3 cycles and last 2 Example 2.7 Some examples of tokenizing a hypothetical segment with index 0 and 9 cycles.
Complex segments with knit
Each element in the knit array is made of one or more token references separated by the : concatenation operator. A token reference is the zero based index of the token pattern in the adjacent token array. Appending the left hand side reverse complementarity ~ operator will instead concatenate the reverse complemented sequence of the token. NOTE Each token reference is evaluated before concatenation so ~ evaluation precedes : evaluation.
Pattern Description 0The simplest possible pattern will assemble a segment from just token 0. ~0Assemble a segment from the reverse complemented token 0. 0:1Assemble a segment by concatenating token 0 and token 1 ~0:1Assemble a segment by concatenating the reverse complement of token 0 and token 1. Example 2.8 Several examples of
knitsyntax for constructing new segments.
Notice that a negative token start coordinate is equivalent to the corresponding positive token end coordinate on the reverse complemented strand and vice verse. More formally the token 0:-x:-y is equivalent to 0:y:x if applied to the reverse complement of the segment. For instance to concatenate to the first output segment the first 6 bases of the reverse complemented strand of the first input segment you would define token 0:-6: and then reference it in the transform pattern for output segment 0 as ~0.
Different context for a transform
A transform declared in the template directive of the instruction assembles the output read and implicitly defines the output segment cardinality: the number of segments in the output read. When declared inside a barcode decoder directive a transform constructs the segmented sequence that is matched against the possible barcode values.
{ "input": [ "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram" ], "template": { "transform": { "token": [ "0:6:", "3::-6" ] } }, "sample": { "transform": { "token": [ "1::8", "2::8" ] } }, "molecular": [ { "transform": { "token": [ "0::6", "3:-6:" ], "knit": [ "4", "~5" ] } } ] }Example 2.9 For a more involved example let us consider that the input we declared in Example 2.2, is a dual indexed paired end read with two 8 cycle multiplex barcode segments on the second and third input segments and two molecular barcode segments, one on the first 6 cycles of the first segment and another, reverse complemented, on the last 6 cycles of the last segment. We extract both molecular barcode segments and remove them from the output segments.
The output directive
The output directive is an ordered array of output file paths. If the output directive contains the same number of file paths as the output segment cardinality (defined by the output directive in template) each segment will be written to the corresponding enumerated output file. If only one file path is declared all segments will be interleaved into that file. Declaring a number of files paths different than 1 or output segment cardinality is ambiguous and will result in a validation error. Read segments are guaranteed to be written contiguously and in-order. The order of the reads in the output, however, is not guaranteed to be consistent with their order in the input or to be stable between successive executions when the thread directive is bigger than 1. If omitted, the output directive defaults to interleaved SAM written to standard output. The same output directive can be spicified inside individual barcode decleartion inside one of the barcode decoder directives to allow splitting reads into different files by the barcode they were classified to. output directives deeper in the JSON tree take precedence over the one declared in the root, so consider the output in the instruction root to be the default unless otherwise specified.
Only one decoder can control output splitting by setting multiplexing classifier to true in the decoder dictionary. In the event of multiple candidates, to resolve which decoder controls output splitting, Pheniqs employs the following search algorithm: If only one decoder has multiplexing classifier set to true it is selected. If more than one has multiplexing classifier set to true a validation error is raised. If none has multiplexing classifier set to true a similar search is performed for a decoder that mentions output. If only one decoder contains an output directives it is selected. If more than one decoder contains an output directives a validation error is raised. if none contains an output directive the sample decoder is selected.
Barcode decoding
Pheniqs can populate the related standardized SAM auxiliary tags for sample, molecular and cellular barcodes, adhering to the recommendations outlined in the SAM specification. We distinguish between closed class decoding algorithms, when a discrete list of classes (in our case nucleotide sequences) is known in advance and so a prior distribution is available, and open class decoders, when the discrete list of classes is unknown and so the prior distribution is hidden. The decoder codec directive specifies the allowed classes for closed class decoders as a JSON dictionary. The keys of the dictionary must be unique strings within the dictionary and play a role in the inheritance model but you may otherwise choose them as you see fit. One simple methodology is to use the concatenated barcode sequence prefixed by an @ sign (to remind you Pheniqs does not actually interpret it as a nucleotide sequence), but you may choose more meaningful names to make your instruction files more readable.
Pheniqs offers a choice of two closed class decoding strategies: the widespread minimum distance decoder (MDD) and a more refined probabilistic Phred-adjusted maximum likelihood decoder (PAMLD). Unlike MDD, PAMLD consults base calling quality scores and the prior barcode distribution and will outperform MDD in almost every real world scenario. Since PAMLD computes the full Bayesian posterior decoding probability to pick the maximum likelihood, the probability of an incorrect barcode assignment is made available in SAM auxiliary tags reserved for local use, for downstream analysis consideration.
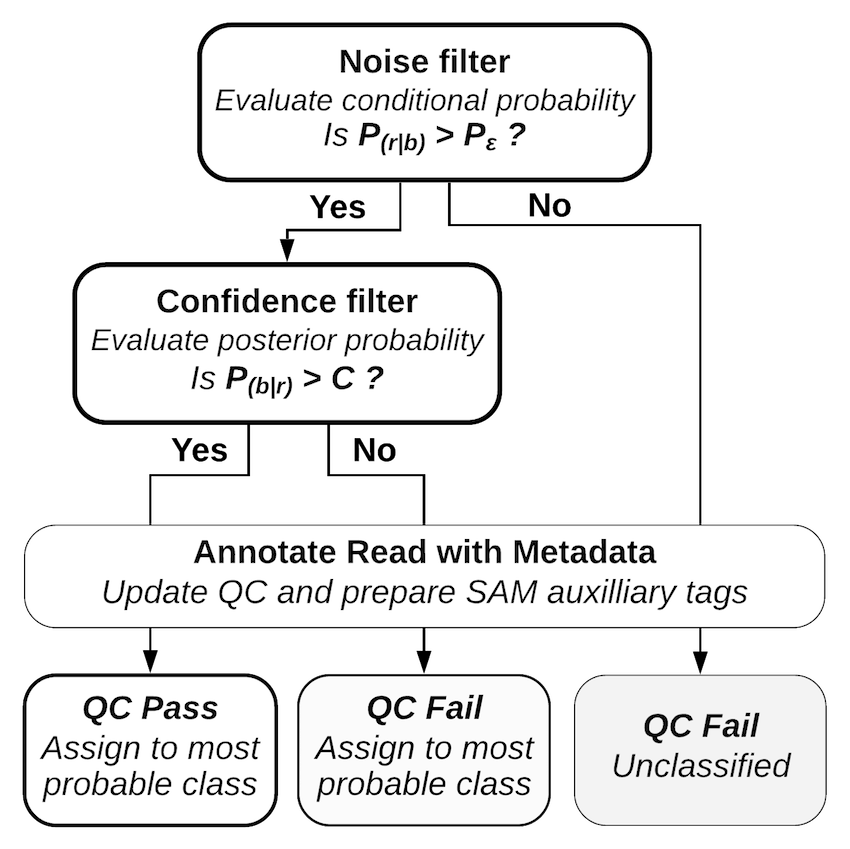
Decoding a read with PAMLD Reads with a lower conditional probability than random sequences fail the noise filter and are classified as noise without further consideration. Reads with a posterior probability that does not meet the confidence threshold fail the confidence filter; these reads are classified, but they are marked as “qc fail” so the confidence threshold can be reconsidered at alater stage. A full description of the mathematics behind Pheniqs, as well as performance evaluations and comparisons with other decoding methods, may be found in the paper. Pheniqs is designed to accommodate the addition of alternative decoders, which can be added as derived classes of a generic decoder object.
Pheniqs currently does not support degenerate IUPAC bases in closed class barcode declarations, but the probabilistic model underlaying PAMLD can be extended to support them if a demand arises.
Since error handling is much trickier in open class decoding, Pheniqs currently only supports a naive decoder that will simply populate the relevant raw, uncorrected, SAM auxiliary tags without attempting error correction. In the case of decoding open class molecular barcodes, all reads associated with a unique molecular identifier are expected to be tagging the same DNA molecule. For that reason, most published error correction strategies involve examining the corresponding biological sequence and aligning it to a consensus sequence of a group of reads that have already been associated with that barcode. A specialized open class error correcting molecular barcode decoder is planned for a future release.
When declaring a decoder directive you specify the decoding strategy by setting the algorithm attribute.
Handling decoding failure
Reads that had one of their sample, cellular or molecular barcodes fail decoding will be marked by Pheniqs as QC Failed. The undetermined decoder tag can be used to inform a decoder how to handle reads that fail decoding. If omitted the implicit undetermined directive inherits global or decoder attributes in the same way that specific barcode classes do. When populating corrected nucleotide and quality sequences in SAM auxiliary tags, a placeholder sequence of = character of the correct layout is used to denote a decoding failure.
Phred-adjusted maximum likelihood decoding
Pheniqs implements a probabilistic closed class phred-adjusted maximum likelihood decoder that directly estimates the decoding likelihood from the base calling error probabilities provided by the sequencing platform. When output is SAM encoded Pheniqs will write the decoding error probability to the XB auxiliary tag for a sample barcode, the XM tag for a molecular barcode and the XC tag for a cellular barcode. In the case of a decoding failure no error probability is reported.
The dominant parameter to set when decoding with PAMLD is the confidence threshold, which is a lower bound on the decoding probability for the decoder to declare a successful classification. The value of confidence threshold defaults to 0.95 but in practice depends on the application and controls the tradeoff between false positives and false negatives. Since the decoding error probability is written to the a SAM auxiliary tag you may choose to set confidence threshold to a relatively lax value and leave the decision to exclude certain reads for downstream analysis.
PAMLD also consults user provided prior barcode instance and decoding failure frequencies. Barcode frequencies are specified in the class concentration attribute while the expected decoding failure frequency is specified in the decoder noise attribute. For the Illumina platform the most common value for noise will be at least the amount of PhiX Control Library spiked into the pooled solution, which is usually 1% and corresponds to a noise value of 0.01. Assays with low base diversity sometimes use a higher concentration to compensate and some applications can go as high as 50%. If you know a priori that some amount of sequences present in the pool represent contamination or multiplexed libraries you are not declaring in the configuration, their concentration should be factored into this parameter. The priors you specify can significantly affect decoding precision. Pheniqs provides a python script to help you estimate the priors from the report emitted by a preliminary run.
The concentration attribute defaults to 1 if omitted. The values for all barcode instances in a decoder are normalized so that their sum equals 1.0 - noise. Notice that unlike concentration the noise attribute is specified as a probability value between 0 and 1, and it rarely make sense to set it to 0. If the concentration attribute is omitted from all classes the result is an implicit uniformly distributed prior.
Minimum distance decoding
Pheniqs also implements the more traditional minimum distance decoder that consults the edit distance between the expected and observed sequence. MDD consults the distance tolerance tag, which is an array of upper bounds on the edit distance between each segment of the expected and observed barcode to still be considered a match. Setting this property to a value higher than the Shannon bound, which also serves as the default value for distance tolerance, can lead to ambiguous classification and will result in a validation error.
Since MDD effectively ignores the Phred encoded quality scores, it may be consulting extremely unreliable base calls. To mitigate that effect you may set the quality masking threshold attribute, which is a lower bound on the permissible base calling quality. Observed bases with quality lower than this threshold will be considered as N by the minimum distance decoder. quality masking threshold defaults to 0 which effectively disables quality masking.
The sample directive
The sample directive accommodates one sample decoder. When decoding the sample barcode Pheniqs will write the raw uncorrected nucleotide barcode sequence to the BC SAM auxiliary tag and the corresponding Phred encoded quality sequence to the QT tag. When decoding with PAMLD, the decoding error probability is written to the XB tag. Sample barcodes classify the read to a read group by populating the RG SAM auxiliary tag, which is a reference to the ID attribute of a read group declared in the SAM header.
{ "input": [ "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram" ], "sample": { "transform": { "token": [ "1::8", "2::8" ] }, "codec": { "@AAGAGGCAAGAGGATA": { "barcode": [ "AAGAGGCA", "AGAGGATA" ] }, "@AGGCAGAAAGAGGATA": { "barcode": [ "AGGCAGAA", "AGAGGATA" ] }, "@CAGAGAGGAGAGGATA": { "barcode": [ "CAGAGAGG", "AGAGGATA" ] }, "@CGTACTAGTCTTACGC": { "barcode": [ "CGTACTAG", "TCTTACGC" ] } } } }Example 2.10 Expanding Example 2.9 we declare 4 classes for a sample decoder which will be be matched against the segmented sequence extracted by the
transform. NOTE: The keys of thecodecdictionary directive have no special meaning and you may choose them as you see fit, as long as they are unique within thecodecdictionary. As a matter of convention we use the concatenation of all barcode segments for each respective class.
Read groups
Sample barcodes are traditionally mapped to the SAM concept of Read groups. In addition to the correct sequence identifying the read group, the SAM RG header tag can contain additional metadata fields that you can either specify for an individual read group or globally for inclusion in all read groups.
{ "flowcell id": "HK5NHBGXX", "flowcell lane number": 1, "input": [ "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram", "HK5NHBGXX_l01.cram" ], "sample": { "CN": "NYU CGSB", "DT": "2016-07-15T07:00:00+00:00", "PI": "500", "PL": "ILLUMINA", "PM": "HiSeq 2500", "transform": { "token": [ "1::8", "2::8" ] }, "codec": { "@AAGAGGCAAGAGGATA": { "barcode": [ "AAGAGGCA", "AGAGGATA" ], "SM": "c57bl6 mouse 1", "LB": "pre b fetal liver technical replicant 1" }, "@AGGCAGAAAGAGGATA": { "barcode": [ "AGGCAGAA", "AGAGGATA" ], "SM": "c57bl6 mouse 1", "LB": "pre b fetal liver technical replicant 2" }, "@CAGAGAGGAGAGGATA": { "barcode": [ "CAGAGAGG", "AGAGGATA" ], "SM": "c57bl6 mouse 1", "LB": "pre b fetal liver technical replicant 3" }, "@CGTACTAGTCTTACGC": { "barcode": [ "CGTACTAG", "TCTTACGC" ], "SM": "c57bl6 mouse 2", "LB": "follicular spleen technical replicant 1", "PI": "300" } } } }Example 2.11 Further expanding Example 2.10 with attributes related the read group SAM header tags. Attributes declared in the decoder will apply to all barcode entries in the
codecdictionary unless explicitly overridden in the entry. For instance all except @CGTACTAGTCTTACGC, that locally overrides PI to be 300, will have their PI tag set to 500.
The SAM specification outlines some attributes associated with a read group.
| Name | Description | Type |
|---|---|---|
| ID | Read group identifier | string |
| BC | Correct barcode sequence | string |
| LB | Library name | string |
| SM | Sample name | string |
| PU | Platform unit unique identifier | string |
| CN | Name of sequencing center producing the read | string |
| DS | Description | string |
| DT | ISO8601 date the run was produced | string |
| FO | Flow order | string |
| KS | Key sequence | string |
| PI | Predicted median insert size | string |
| PL | Platform or technology used to produce the reads | string |
| PM | Platform model | string |
| PG | Programs used for processing the read group | string |
- ID defaults to the value of PU if not explicitly specified. If explicitly declared it must be unique within the
codecdictionary. - PU, following the convention established by GATK, defaults to
flowcell id:flowcell lane number:barcode. Ifflowcell idorflowcell lane numberare not specified they are omitted along with their trailing:. If explicitly declared PU must be unique within thecodecdictionary. - PL, as defined in the SAM specification, is one of CAPILLARY, LS454, ILLUMINA, SOLID, HELICOS, IONTORRENT, ONT, PACBIO.
- BC is computed from the declared barcode seuqnce according to SAM conventions.
The molecular directive
The molecular directive can be used to declare an array containing multiple decoders. When decoding molecular barcodes Pheniqs will write the nucleotide barcode sequence to the RX SAM auxiliary tag and the corresponding Phred encoded quality sequence to the OX tag. A molecular identifier is written to the MI tag.
If error correction is applied to the barcode the raw, uncorrected, nucleotide, and quality sequences are written to the QX and BZ tags and the decoding error probability to the XM tag.
The cellular directive
The cellular directive can be used to declare an array containing multiple decoders. When decoding cellular barcodes Pheniqs will write the raw, uncorrected, nucleotide barcode sequence to the CR SAM auxiliary tag and the corresponding Phred encoded quality sequence to the CY tag, while The decoded cellular barcode is written to the CB tag. The decoding error probability is written to the XC tag.
URL handling
Setting global URL prefixes can make your instruction file more portable. If specified, the base input url and base output url are used as a prefix to relative URLs defined in the input and output directives, respectively. A URL is considered relative if it does not begin with a / character. Environment variables in URLs will be resolved by Pheniqs when your instruction file is compiled. base input url and base output url default to the working directory which is the directory where Pheniqs was executed, so if the a corresponding base directory is not specified, relative URLs are resolved against the working directory.
{ "base input path": "/volume/alpha/HK5NHBGXX", "input": [ "HK5NHBGXX_S1_L001_R1_001.fastq.gz", "HK5NHBGXX_S1_L001_I1_001.fastq.gz", "HK5NHBGXX_S1_L001_I2_001.fastq.gz", "HK5NHBGXX_S1_L001_R2_001.fastq.gz", ] }Example 2.12 Providing a
base input pathandbase output pathand relative URLs in theinputoroutputdirectives is a good way to make your instruction file more portable.
{ "input": [ "/volume/alpha/HK5NHBGXX/HK5NHBGXX_S1_L001_R1_001.fastq.gz", "/volume/alpha/HK5NHBGXX/HK5NHBGXX_S1_L001_I1_001.fastq.gz", "/volume/alpha/HK5NHBGXX/HK5NHBGXX_S1_L001_I2_001.fastq.gz", "/volume/alpha/HK5NHBGXX/HK5NHBGXX_S1_L001_R2_001.fastq.gz", ] }Example 2.13 Compiling the directives in Example 2.12.
{ "base input path": "~/HK5NHBGXX", "input": [ "HK5NHBGXX_S1_L001_R1_001.fastq.gz", "HK5NHBGXX_S1_L001_I1_001.fastq.gz", "HK5NHBGXX_S1_L001_I2_001.fastq.gz", "HK5NHBGXX_S1_L001_R2_001.fastq.gz", ] }Example 2.14
base input pathandbase output pathdo not have to be absolute URLs and may contain environment variables enclosed in curly brackets. The~character in the beginning of a URL is interpreted as theHOMEenvironment variable on most POSIX shells and resolves to the home directory of the currently logged in user. relative URLs are resolved against theworking directorywhich is the directory where Pheniqs was executed.
Query Parameters
Pheniqs will try to guess output format and compression from the output URL file extensions. You can however specify those explicitly using URL query parameter. Parameters explicitly specified in the query will override guessed parameters. In some cases a query parameter is the only way to specify those values because the file name is uninformative, for instance when writing to standrd output.
| Name | Description | Type |
|---|---|---|
| format | file format | sam, bam, cram or fastq |
| compression | compression algorithm for fastq and bam formats |
gz, bgzf, none |
| level | compression level for fastq, bam and cram formats |
0-9 |
Standard streams
If the input directive is omitted Pheniqs will expect input on /dev/stdin. If the output directive is omitted Pheniqs will write SAM to /dev/stdout. Regardless of file extensions, Pheniqs will detect input format by inspecting the first few bytes of your input and will attempt to guess the input resolution and layout. When writing to /dev/stdout you might want to explicitly provide a format with the format, compression and level URL query parameter. Alternatively you can specify defaults with -F/--format, -Z/--compression and -L/--level command line arguments, but those will apply to all otherwise unspecified files. You may even specify /dev/null as a URL in an output directive to discard the output.
{ "output": [ "/dev/stdout?format=cram" ] }Example 2.15 Declaring interleaved CRAM output to standard output using the format URL query parameter.
Phred offset
The input phred offset and output phred offset are applicable only to FASTQ files and specify the Phred scale decoding and encoding offset, respectively. The default value for both is 33, knowns as the Sanger format. The binary HTSlib formats BAM and CRAM encode the quality value numerically and so require no further manipulation. The sequence alignment map format specification states that text encoded SAM records always use the Sanger format.
{ "output phred offset": 33, "input phred offset": 33 }Example 2.16 Both the
input phred offsetandoutput phred offsetdefault to 33, known as the Sanger format.
Leading Segment
Pheniqs constructs new segments for the output read and must generate corresponding identifiers and replicate metadata from the input. Since segments can potentially disagree on metadata, one input segment is elected as the leader and used as a template when constructing the output segments. The leading segment property is a reference to an input segment index and defaults to 0 if omitted.
{ "leading segment": 0 }Example 2.17 The leading segment defaults to be the first input segment.
Pass filter / QC fail reads
Some input reads are marked as failing quality control. For instance, Illumina sequencers perform an internal quality filtering procedure called chastity filter, and only reads that pass this vendor quality control filter are marked as pass-filter. Pheniqs marks reads that have failed barcode decoding as QC fail. Setting the filter incoming qc fail configuration attribute to true, or specifying the -N/--no-input-npf command line argument, will instruct Pheniqs to disregard incoming reads marked as not passing filter. This allows to filter reads that have been marked as QC fail by a previous processing tool. Setting the filter outgoing qc fail attribute to true, or specifying the -n/--no-output-npf command line argument, will instruct Pheniqs not to include reads marked as not passing filter in the output. That will filter incoming QC fail (if they have not already been filtered with filter incoming qc fail) as well as reads that have been marked by Pheniqs as QC fail during barcode decoding.
Configuration validation and compliation
The -V/--validate command line flag makes Pheniqs evaluate the supplied instruction and emit a human readable description of the instruction without actually executing it. It is sometimes useful to inspect this description before executing to make sure all implicit parameters are allocated the desired values. To also print out the barcode distance metric for each closed class decoder you may additionally set the -D/--distance command line flag. The top half of the matrix, above the diagonal, is the pairwise Hamming distance, while the bottom half is the maximum number of correctable errors the pair can tolerate, known as the Shannon bound.
Quality Control and Statistics
Pheniqs emits a statistical report. If you specify the -q/--quality command line flag the report will also include a comprehensive quality control report.
Decoder statistics
For every sample, molecular and cellular decoder the following statistics is reported, if applicable. Counters have a slightly different meaning depending on the value of the filter incoming qc fail flag. if filter incoming qc fail was true incoming reads are dropped immediately and are never seen by the decoder and so counters will only be affected by reads that were marked as QC fail by Pheniqs due to barcode decoding failure. If filter incoming qc fail was false the decoder will attempt to classify all reads and so counters will also count reads that were already marked as QC fail in the input. pf counters will always only apply to reads that were either marked QC fail by Pheniqs nor were already marked QC fail in the input.
| JSON field | Description |
|---|---|
| count | count for all reads processed by the pipeline, both classified and unclassified. |
| classified count | count of all reads classified to some barcode. |
| classified fraction | classified count / count |
| average classified distance | average hamming distance when decoding classified reads. |
| average classified confidence | average confidence when decoding classified reads. |
| pf count | count of output reads processed by the pipeline that passed quality control. |
| pf fraction | pf count / count |
| pf classified count | count of all output reads classified to some barcode that passed quality control. |
| pf classified fraction | pf classified count / pf count |
| classified pf fraction | pf classified count / classified count |
| average pf classified distance | average hamming distance of output classified reads that passed quality control. |
| average pf classified confidence | average confidence of classified output reads that passed vendor quality control. |
| low conditional confidence count | count of reads that failed to classify due to low conditional confidence. |
| low confidence count | count of reads that failed to classify due to low confidence. |
| estimated noise | estimated ratio of noise reads. |
Barcode statistics
In every decoder statistics the unclassified element reports statistics about reads that failed to be classified, while each element in the classified array reports statistics for one of the barcodes.
The decoder:: prefix in the table refers to the attribute in the parent decoder statistics.
| JSON field | Description |
|---|---|
| count | count of reads classified to the barcode. |
| average distance | average hamming distance between observed and decoded barcode. |
| average confidence | average confidence of decoding a barcode. |
| pooled fraction | count / decoder::count |
| pooled classified fraction | count / decoder::classified count |
| pf count | count of reads classified to the barcode that passed quality control. |
| pf fraction | pf count / count |
| average pf distance | average hamming distance between observed and decoded barcode in reads that passed quality control. |
| average pf confidence | average confidence of decoding a barcode in reads that passed quality control. |
| pf pooled fraction | pf count / decoder::pf count |
| pf pooled classified fraction | pf count / decoder::pf classified count |
| low conditional confidence count | count of reads that failed to classify due to low conditional confidence. |
| low confidence count | count of reads that failed to classify due to low confidence. |
| estimated concentration | estimated ratio of reads classified to the barcode. |
Prior estimation
The statistical report Pheniqs produces when decoding with PAMLD can be used to estimate concentration and noise from the data. The mathematical procedure itself, as well as the parameters involved, are described in the PAMLD page. The --prior command line parameter, if specified, is a path where a new JSON encoded configuration file will be written that contains the same decoding job executed but with concentration and noise values adjusted based on the statistics collected from the run. To use it, simply execute Pheniqs again with the new configuration file. The initial priors can be either left blank, suggesting a uniform prior, or your best guess.
{ "sample": { "average classified confidence": 0.9999766864884027, "average classified distance": 0.01468791896320572, "average pf classified confidence": 0.999980549273765, "average pf classified distance": 0.00811558561328005, "classified": [ { "CN": "CGSB", "DT": "2018-02-25T07:00:00+00:00", "ID": "BDGGG:AGGCAGAA", "LB": "trinidad 5", "PI": "300", "PL": "ILLUMINA", "PM": "miseq", "PU": "BDGGG:AGGCAGAA", "SM": "trinidad", "average confidence": 0.9999677414000366, "average distance": 0.01726894787336105, "average pf confidence": 0.9999696078665885, "average pf distance": 0.010310965630114567, "barcode": [ "AGGCAGAA" ], "concentration": 0.20357142857142858, "count": 6254, "index": 1, "pf count": 6110, "pf fraction": 0.9769747361688519, "pf pooled classified fraction": 0.18782662158007994, "pf pooled fraction": 0.18343390675192892, "pooled classified fraction": 0.18632504096529124, "pooled fraction": 0.18180761068635718 } ], "classified count": 33565, "classified fraction": 0.97575510916015, "classified pf fraction": 0.96916430805899, "count": 34399, "pf classified count": 32530, "pf classified fraction": 0.976612927437029, "pf count": 33309, "pf fraction": 0.9683130323555917, "unclassified": { "CN": "CGSB", "DT": "2018-02-25T07:00:00+00:00", "ID": "BDGGG:undetermined", "PI": "300", "PL": "ILLUMINA", "PM": "miseq", "PU": "BDGGG:undetermined", "SM": "trinidad", "count": 834, "index": 0, "pf count": 779, "pf fraction": 0.934052757793765, "pf pooled classified fraction": 0.023947125730095298, "pf pooled fraction": 0.02338707256297097, "pooled classified fraction": 0.024847311187248624, "pooled fraction": 0.024244890839849998 } } }Example 2.18 Partial example of a sample decoder statistics report
Quality Control
{ "sample": { "classified count": 0, "classified fraction": 0.0, "classified pf fraction": 0.0, "count": 34399, "pf classified count": 0, "pf classified fraction": 0.0, "pf count": 33309, "pf fraction": 0.9683130323555917, "unclassified": { "CN": "CGSB", "DT": "2018-02-25T07:00:00+00:00", "ID": "BDGGG:undetermined", "PI": "300", "PL": "ILLUMINA", "PM": "miseq", "PU": "BDGGG:undetermined", "SM": "trinidad", "count": 34399, "index": 0, "pf count": 33309, "pf fraction": 0.9683130323555917, "pf pooled fraction": 1.0, "pooled fraction": 1.0, "quality control by segment": [ { "average phred score report": { "average phred score distribution": [ 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 56, 75, 100, 146, 115, 99, 95, 71, 74, 69, 65, 45, 48, 81, 69, 58, 53, 76, 113, 112, 153, 225, 340, 883, 31178, 0, 0, 0, 0, 0, 0 ], "average phred score max": 35.5, "average phred score mean": 34.69170978807524, "average phred score min": 0.0 }, "max sequence length": 8, "min sequence length": 8, "quality control by cycle": { "cycle quality distribution": { "cycle count": [ 34399, 34399, 34399, 34399, 34399, 34399, 34399, 34399 ], "cycle quality first quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality interquartile range": [ 0, 0, 0, 0, 0, 0, 0, 0 ], "cycle quality left whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality max": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality mean": [ 33.19076135934184, 33.330213087589758, 33.32413732957353, 33.33056193493997, 33.32210238669729, 37.09950870664845, 37.06901363411727, 36.86737986569377 ], "cycle quality median": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality min": [ 12, 12, 12, 12, 12, 10, 10, 10 ], "cycle quality right whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality third quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ] } }, "quality control by nucleotide": [ { "cycle quality distribution": { "cycle count": [ 6397, 7845, 15238, 6792, 6444, 5988, 13037, 14189 ], "cycle quality first quartile": [ 34, 34, 34, 34, 34, 38, 38, 37 ], "cycle quality interquartile range": [ 0, 0, 0, 0, 0, 0, 0, 1 ], "cycle quality left whisker": [ 34, 34, 34, 34, 34, 38, 38, 35 ], "cycle quality max": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality mean": [ 32.93950289198062, 33.33142128744423, 33.456227851424078, 33.05005889281507, 33.08628181253879, 36.72227788911156, 37.06251438214313, 36.85340756924378 ], "cycle quality median": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality min": [ 12, 12, 12, 12, 12, 10, 10, 10 ], "cycle quality right whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality third quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ] }, "nucleotide": "A", "nucleotide count": 75930 }, { "cycle quality distribution": { "cycle count": [ 6798, 5944, 6034, 13875, 6802, 15286, 7626, 5989 ], "cycle quality first quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality interquartile range": [ 0, 0, 0, 0, 0, 0, 0, 0 ], "cycle quality left whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality max": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality mean": [ 33.03147984701383, 33.1771534320323, 33.09247596950613, 33.431423423423428, 33.279917671273157, 37.35313358628811, 36.97521636506688, 36.64601769911504 ], "cycle quality median": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality min": [ 12, 12, 12, 12, 12, 10, 10, 10 ], "cycle quality right whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality third quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ] }, "nucleotide": "C", "nucleotide count": 68354 }, { "cycle quality distribution": { "cycle count": [ 7410, 20367, 6345, 7765, 13446, 6370, 13500, 6616 ], "cycle quality first quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality interquartile range": [ 0, 0, 0, 0, 0, 0, 0, 0 ], "cycle quality left whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality max": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality mean": [ 33.35681511470985, 33.585162272303239, 33.3117415287628, 33.511912427559568, 33.62152312955526, 37.0265306122449, 37.4762962962963, 37.18636638452237 ], "cycle quality median": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality min": [ 12, 12, 12, 12, 12, 10, 10, 10 ], "cycle quality right whisker": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality third quartile": [ 34, 34, 34, 34, 34, 38, 38, 38 ] }, "nucleotide": "G", "nucleotide count": 81819 }, { "cycle quality distribution": { "cycle count": [ 13794, 243, 6782, 5967, 7707, 6755, 236, 7605 ], "cycle quality first quartile": [ 34, 12, 34, 34, 34, 38, 11, 38 ], "cycle quality interquartile range": [ 0, 0, 0, 0, 0, 0, 13, 0 ], "cycle quality left whisker": [ 34, 12, 34, 34, 34, 38, 10, 38 ], "cycle quality max": [ 34, 34, 34, 34, 34, 38, 38, 38 ], "cycle quality mean": [ 33.29657822241554, 15.666666666666666, 33.24506045414332, 33.179319591084297, 33.03412482159076, 36.92879348630644, 17.161016949152545, 36.79026955950033 ], "cycle quality median": [ 34, 12, 34, 34, 34, 38, 11, 38 ], "cycle quality min": [ 12, 12, 12, 12, 12, 10, 10, 10 ], "cycle quality right whisker": [ 34, 12, 34, 34, 34, 38, 38, 38 ], "cycle quality third quartile": [ 34, 12, 34, 34, 34, 38, 24, 38 ] }, "nucleotide": "T", "nucleotide count": 49089 } ], "url": "/dev/null" } ] } } }Example 2.19 Partial example of a multiplex decoder statistics report with QC